Polypoidal Choroidal Vasculopathy
Gisela Barcat Angelelli
D. Virgil Alfaro III
Lawrence Yannuzzi
Richard J. Antcliff
INTRODUCTION HISTORY
Polypoidal choroidal vasculopathy (PCV) is a distinct clinical entity characterized by a branching choroidal vascular network with polypoidal vascular dilations at the border of that network (1).
It was first described by Yannuzzi et al. (2) as a variant of choroidal neovascularization (CNV) in African American women in the peripapillary area, and they proposed the term “idiopathic polypoidal choroidal vasculopathy.”
Kleiner et al. (3) later described a similar group of patients in a presentation entitled “Posterior Uveal Bleeding Syndrome.”
Subsequently Stern et al. (4) described recurrent, multiple, bilateral, asymmetric, serosanguineous retinal pigment epithelium (RPE) detachments in a group of black women.
Yannuzzi et al. described these vessels as a distinct abnormality of the choroidal vasculature with two demonstrable components: (i) dilated and branching inner choroidal vessels, and (ii) terminal reddish-orange, spheroid aneurysmal-like, definitions which were called “polyp-like” (1).
This new clinical description led to Yannuzzi and collaborators dropping the word “idiopathic” and introducing the term polypoidal choroidal vasculopathy or PCV in 1990 (1).
Since then, with the advent of diagnostic techniques, such as indocyanine green angiography (ICGA), the spectrum of the vasculopathy has been better illustrated by the longer wavelengths used to excite the indocyanine molecule, providing a more detailed representation of choroidal circulation (5).
Knowledge and understanding of PCV has increased over the last two decades, but there are still some pathogenic mechanisms that need further study.
This chapter focuses on providing a guide to clinical manifestations, diagnosis, and treatment strategies for patients with PCV based on the findings of clinical trials.
EPIDEMIOLOGY
In pigmented races, such as African Americans and Asians, PCV is the main vascular component of neovascular maculopathies.
Also known to occur in white-race patients with or without concomitant drusen, this form of neovascularization is characteristically distributed from the peripapillary area to the peripheral fundus.
The age of diagnosis can range from the 20s to the 80s, but PCV is most commonly diagnosed between 60 and 70 years of age (6).
In Asian races, PCV is the most common form of macular disease in adults and elderly people (7,8), including Korean (9) (mean age 64.6 years), Chinese (10) (mean age 65.4 years), and Japanese (11) people (mean age 72.8 years).
Initially PCV had been thought to be a condition exclusively found in women (4). Subsequent studies have shown that while there is a female preponderance in Caucasian races (75%), in Asian races there is a male preponderance (71%).
PCV can affect both eyes, having a considerable likelihood of developing lesions in a fellow eye. Several studies in Asian and European populations showed that the incidence of bilateral lesions is 24.1% in Koreans (9), 18.4% in Japanese (11), 24.7% (12) in Chinese (10), and 25% in Europeans (13).
The typical soft drusen of age-related macular degeneration (AMD) are not usually present particularly in highly pigmented individuals, but they may be seen in white patients. Furthermore, their distribution is generally in the central macula area in highly pigmented individuals, while Caucasian patients more typically have involvement in the peripapillary area.
Yannuzzi et al. (14) showed that drusen were observed in 63 (70%) of 90 fellow eyes with unilateral macular degeneration compared with only one (16.7%) of six fellow eyes with polypoidal CNV. Among Japanese patients with PCV, 23% showed soft drusen in the central macula in conjunction with the polypoidal vascular abnormality (15).
The distribution of the lesions also appears to be different in pigmented and Caucasian races. While in African Americans and Asians the lesions are generally in the macular area, in Caucasians these lesions are more typically located in the peripapillary area (13).
GENETICS AND BIOMARKERS
Genetic studies suggest that PCV is a type of CNV. HTRA1 is a membrane serine peptidase 1 found to be associated with both neovascular AMD (12,18) and PCV (19,20,21).
This gene is thought to regulate degradation of extracellular matrix proteoglycan. Overexpression of HTRA1 caused by a single-nucleotide mutation may alter the integrity of Bruch’s membrane, and facilitate the expansion of abnormal choroidal capillaries (20).
Furthermore, both in Asian and white populations, PCV has been shown to share certain risk and protective alleles related to the complement system. Polymorphism for genes of complement factor H (CFH), complement component 2, and factor B were investigated in PCV, and a significant association was noted with CFH variants rs3753394 and rs800292 (6,22).
On the other hand, the Y204H variant of CFH (rs1061170) and the BF and C2 variants were not significantly associated with PCV (23).
Coding variant I62V in the CFH gene (24) and the variants of ARMS2 (LOC387715), a gene coding mitochondrial protein in photoreceptors (25,26), were found to be associated in Japanese patients with PCV (19).
Another study using a Japanese population showed that there was no significant difference in the incidence of CFH Y402H and HTRA rs11200638 between eyes with typical exudative AMD and with PCV (27).
Recent genetic studies have demonstrated an association between genotype and outcomes in the treatment of PCV. Polyp regression and resolution of leakage were found to be significantly better in particular genotypes of the LOC387715 and HTRA1 genes following combination therapy with verteporfin PDT and bevacizumab (28,29).
CLINICAL MANIFESTATIONS
The principal clinical manifestations of PCV are seen in the posterior segment. The presence of orange-red nodule-like structures beneath the RPE associated with an adjacent serous pigment epithelial detachment or overlying neurosensory detachment (Fig. 7.1A), subretinal hemorrhage (Fig. 7.1B), and lipid exudates (Fig. 7.1C) is considered the clinical hallmark of PCV. In some patients, progressive subretinal fibrosis and atrophic scarring can be observed (Fig. 7.1D).
PCV has been classified clinically (30) as (i) quiescent, polyps in the absence of subretinal or intraretinal fluid or hemorrhage; (ii) exudative, exudation without hemorrhage, which includes variously neurosensory retinal thickening, neurosensory detachment, PED, and subretinal lipid exudation; and (iii) hemorrhagic, any subretinal or sub-RPE hemorrhage with or without other exudative characteristics. This clinical classification described the primary features of the disease, but the evidence regarding its correlation with prognosis or treatment selection is limited.
The typical manifestations in a patient who is symptomatic for less than 3 months are extensive subretinal exudation and bleeding with minimal cystic change in the retina and a surprisingly good visual acuity (VA). This discrepancy between the severity of the serosanguineous detachments and good visual acuity is best explained by the minimal intraretinal changes. For patients with symptoms longer than 3 months, there are considerable lipid depositions from protein leakage from active aneurysmal elements in the polypoidal vascular abnormalities (6) (Fig. 7.1C).
The morphology of PCV can be distributed into three groups (i) single polyp (Fig. 7.2A); (ii) cluster: more than two polyps in a group (Fig. 7.2B); and (iii) string: three or more polyps in a line (Fig. 7.2C).
Cackett et al. (31) found cluster formation in 66.7% of the cases, single in 27.5%, and string in 5.8% in a retrospective study of 123 patients.
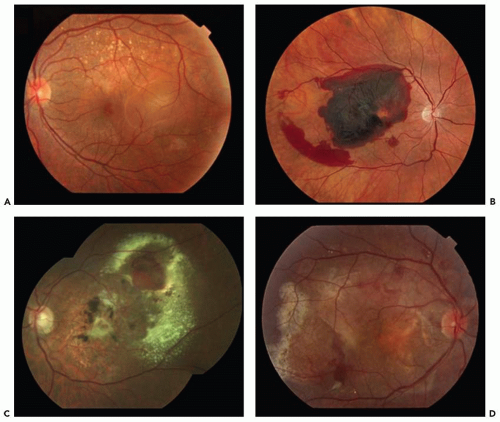 Figure 7.1 ▪ Representative images of polypoidal choroidal vasculopathy (PCV). Pigment epithelial detachment (A) and subretinal hemorrhage (B) are common manifestations. C. Subretinal lipid is often observed. D. In some patients, progressive subretinal fibrosis can be observed. (Reprinted from Imamura Y, Engelbert M, Iida T, et al. Polypoidal choroidal vasculopathy: a review. Surv Ophthalmol. 2010;55(6):501-515. doi: 10.1016/j.survophthal.2010.03.004. Epub 2010 Sep 20, with permission.) |
Uyama et al. (32) found that PCV lesions resembling a cluster of grapes had marked bleeding and leakage and high risk of severe visual loss.
It therefore seems that clusters are associated with an increased frequency of massive hemorrhage resulting in poorer visual outcomes.
NATURAL HISTORY OF THE DISEASE
The natural history of PCV depends on factors including location (peripapillary vs. macular), size of the lesion, and associated bleeding and exudation—which may resolve or progress, sometimes to extensive subretinal fibrosis. The evolution of this condition is probably heavily influenced by the racial background and individual genetic makeup. In Japanese patients, PCV is a chronic disease with a variable course. The risk factors seen in CNV secondary to AMD, including soft confluent drusen and focal hyperpigmentation, are not notable findings in patients with PCV.
Despite having recurrent leakage and bleeding, not all patients develop disciform scars, and others may retain useful vision for years. Some patients, however, experience severe visual loss from a combination of extensive bleeding, exudation, and macular damage.
In a study of the natural history of patients with PCV, Uyama et al. (32) reported that half of the study eyes had
hemorrhagic episodes, recurrent leakage, or severe RPE atrophy after a long follow-up period (24-54 months) (33). Following the spontaneous resolution of the acute serosanguineous complications, there may be signs of subretinal fibrosis, pigment epithelial hyperplasia, and atrophic degeneration (34) (Fig. 7.1D). The incidence of sub-RPE hemorrhage or subretinal hemorrhage described in patients with PCV is high (30%-64%) (35,36).
hemorrhagic episodes, recurrent leakage, or severe RPE atrophy after a long follow-up period (24-54 months) (33). Following the spontaneous resolution of the acute serosanguineous complications, there may be signs of subretinal fibrosis, pigment epithelial hyperplasia, and atrophic degeneration (34) (Fig. 7.1D). The incidence of sub-RPE hemorrhage or subretinal hemorrhage described in patients with PCV is high (30%-64%) (35,36).
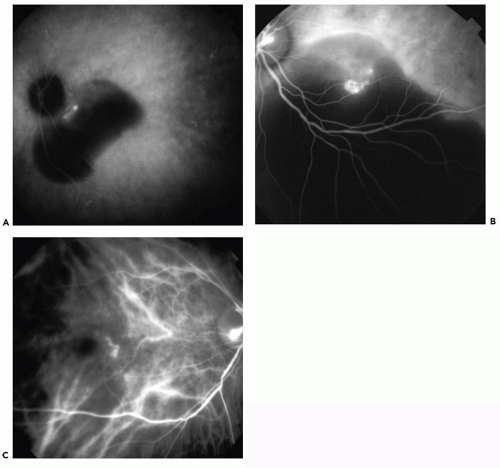 Figure 7.2 ▪ Midphase ICGA revealing: A. Single polyp. B. Cluster of polyps. C. String of polyps. (Reprinted from Cackett P, Wong D, Yeo I. A classification system for polypoidal choroidal vasculopathy. Retina. 2009;29(2):187-191. doi: 10.1097/IAE.0b013e318188c839, with permission.) |
On the other hand, Uyama et al. (32) described stabilization of visual acuity and regression of neovascular lesion with good visual prognosis in 50% of eyes with polypoidal CNV in the posterior pole that were followed for at least 2 years.
PCV has been described in association with other macular abnormalities, such as central serous chorioretinopathy (37,38), typical neovascular (type 1 or 2) AMD (39), sickle cell retinopathy (40), melanocytoma of the optic nerve (41), circumscribed choroidal hemangioma (42), tilted disk syndrome (43), pathological myopia (43), choroidal osteoma (44




Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
