The laryngopharynx coordinates the four basic physiologic functions of the upper aerodigestive tract: airway protection, breathing, swallowing, and phonation. Accurate execution of these tasks depends on proper movement and valving function of the larynx, proper contractile function of the pharynx, and intact sensation of these structures. Neurologic disorders of the laryngopharynx are largely disorders of function (and therefore movement) rather than anatomy and may result from a focal disease process or present as a local manifestation of a systemic disorder. Many diseases produce a stereotypical pattern of abnormal function in the context of historical clues that should be recognized (1).
It is important to understand as an otolaryngologist which disorders are related to end organ dysfunction and which may be due to central dysfunction. It is not unusual for a patient who cannot create normal speech due to a cortical stroke (such as apraxia or dysarthria) to be referred for “hoarseness.” Such an individual may have nothing wrong with his or her larynx or with vocal fold motion or structure but is unable to produce speech output due to cortical disease.
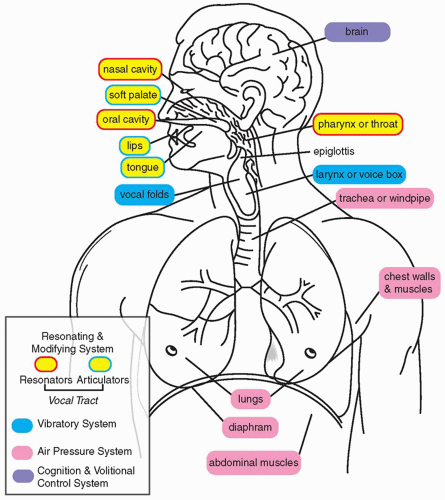
Phonation, articulation, speech, and language are interrelated tasks that require progressively higher orders of brain function (Fig. 70.1). Phonation, defined as the production of sound by the vocal apparatus (usually but not exclusively the vocal folds), is driven by airflow from the lungs. The resultant sound is modified and amplified through resonation within the sinonaso-, oro-, and hypopharyngeal spaces. Articulation further modulates this sound with the palate, tongue, lips, and teeth to produce specific phonemes. Speech requires pronunciation of words from a collection of phonemes, and language involves meaningful groupings of words into phrases to communicate ideas. The mechanics of phonation and articulation are principally influenced by structural defects (such as surgical defects in the tongue or palate), lower motor neuron disorders (vocal fold paralysis), and brainstem function, whereas language requires control by the cerebral cortex.
NEUROANATOMY AND THE LARYNX
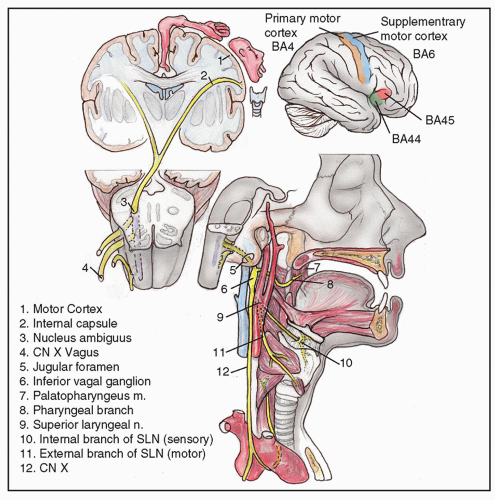
Understanding the implications of neurologic disease of the larynx begins with a working familiarity of the neuroanatomical structures critical to laryngeal function (Fig. 70.2).
Primary motor control of the larynx and receipt of sensory information is accomplished through descending and ascending pathways in the central and peripheral nervous system. The cerebral cortex, divided by Brodmann into a number of areas based on location and function, is the origin of motor commands. The preparatory stages of phonation involve the selection of phonemes in the superior posterior temporal and inferior parietal lobes. Grammatical speech is formulated in Broca’s area (Brodmann areas 44, 45) at the inferior frontal gyrus along the sylvian fissure. This information is communicated to the supplementary motor area in the frontal cortex (Brodmann area 6), which receives inputs from the thalamus (and indirectly from the cerebellum and basal ganglia) for motor modulation, and then to the pyramidal (corticobulbar) neurons in the primary motor cortex (Brodmann area 4). These are the upper motor neurons (UMN) involved in laryngeal control. The processes of these cortical neurons descend through the subcortical white matter to the medial aspect of the anterior midbrain in an area called the cerebral peduncles. These corticobulbar fibers continue their caudal path through the anterior pons where they interact with fibers that originate in the cerebellum, and then a portion cross the midline (decussate), synapsing on the neurons in bilateral nucleus ambiguus of the posterolateral caudal medulla. It is important to repeat that the corticobulbar fibers from one side of the cortex synapse on the neurons of both (bilateral) nucleus ambiguus in the brainstem.
Figure 70.1 The human vocal process. Human vocal communication—effortless for a healthy adult—is the end result of a complex cognitive process leading to a highly coordinated series of motor actions of the thorax, larynx, pharynx, and mouth. The first step is volitional planning of a given vocalization in the cerebrum. The lungs, chest wall, and diaphragm then provide airflow to the vocal apparatus (usually the larynx) that provides a vibration source or a sound source. The resultant sounds are then shaped and modified by the resonating cavities of the upper airway (oral, nasal, and pharyngeal cavities) and by the articulators (palate, lips, teeth, tongue). Disease at any level can lead to a communication deficit. A stroke affects motor planning in the cerebrum, asthma or pulmonary disease can affect the air pressure system, lesions on the vocal cords will affect vibration, and disease of the upper airway or oral cavity can affect articulation or resonation (think of an individual after glossectomy, or an individual with a cleft palate, which changes vocal resonance).
Cell bodies in the nucleus ambiguus are the lower motor neurons and axons (referred to by neuroanatomists as special visceral efferents) for motor control of the larynx through the vagus nerve (cranial nerve X). The vagus exits the anterior brainstem just lateral of midline above the foramen magnum, traversing cerebrospinal fluid, and exiting the skull via the jugular foramen. The vagus travels in the carotid sheath with the carotid artery and jugular vein and gives off the superior laryngeal nerve and the recurrent laryngeal nerves. The superior laryngeal nerve branches medially off the vagus, splitting into the (sensory) internal superior laryngeal nerve, which pierces the thyrohyoid membrane, and the (motor) external superior laryngeal nerve, which descends to innervate the cricothyroideus muscle. The recurrent laryngeal nerve (RLN) branches from the vagus to loop around the arch of the aorta on the left, and around the subclavian artery on the right. The recurrent nerves then ascend in the trachea-esophageal groove posterior to the thyroid gland to innervate all the intrinsic laryngeal musculature save the cricothyroideus.
General sensory information from the larynx mimics the primary motor control pathways largely in reverse. The internal branch of the superior laryngeal nerve provides sensation above the vocal folds, and the RLN provides sensation to the subglottis and proximal trachea. These afferent impulses ascend via the vagus to the bipolar neuronal cell bodies in the inferior vagal ganglion located within the carotid sheath and the jugular foramen to enter the brainstem in the caudal medulla and synapse with neurons in the contralateral spinal trigeminal nucleus. They then ascend in the trigeminothalamic tract to the thalamus, and finally arrive at the primary sensory cortex located in the postcentral gyrus, Brodmann area 3, 1, 2.
The cerebellum and extrapyramidal systems integrate sensory and motor information to exert a modulating influence on laryngeal function. A series of inhibitory and excitatory checks and balances in the basal ganglia allows for secondto- second gradation of laryngeal motor control. The basal ganglia are located in the subcortex of the cerebrum, adjacent to the midbrain and the thalamus. The basal ganglia consist of the globus pallidus, putamen, and caudate. These have further interaction with the substantia nigra located in the anterior midbrain, the subthalamic nucleus, and the thalamus. In general, sensory information arrives from the cortex to the caudate and putamen, and excitatory impulses are sent to the globus pallidus with inhibitory adjustment from the substantia nigra (particularly the dopaminergic pars compacta region). The globus pallidus, with influence from the subthalamic nucleus, exerts a net inhibitory effect on the motor portion of the thalamus, which projects to the frontal planning regions of the cortex. For the cerebellum, sensory information is relayed through the medulla, ultimately projecting to the cerebellar hemispheres and the dentate nucleus, the most lateral deep cerebellar nucleus. Projections are also received from the parietal cortex. These, in turn, are further modulated by inputs from corticobulbar fibers in the pons, and project to the ventral lateral nucleus of the thalamus, which sends inputs back to the motor planning and primary motor areas of the cortex.
Figure 70.2 The pathway of the vagus.
Function follows from anatomy. Motor neurons have two primary purposes, the first of which is establishing control over muscular function, and the second is providing basal tone to those muscles. UMN, originating in the cortex and terminating in the brainstem, have an inhibitory influence on lower motor neurons, acting to reduce tone in effector muscles. Lower motor neurons, in turn, provide the resting tonal stimulation to maintain muscle mass and tension. The “motor unit” consists of the total complement of axons, corresponding neuromuscular junctions, and contractile muscle fibers innervated by a single lower motor neuron. Interruption of the upper and lower motor neuron pathways creates significantly different abnormalities.
A UMN lesion to the corticobulbar tracts (at the contralateral cortex, the subcortical cerebral hemispheres, the cerebral peduncles, or the pons), would create spasticity and increased muscle tone with poor control. The voice would have a variable and unpredictable quality, which would be highly frustrating to the patient. There is not a characteristic laryngoscopic appearance—but the patient may show variable laryngeal motion depending on whether the task is a vegetative task (cough) or volitional task (speech). UMN lesions can be unilateral or bilateral, occur in combination, or in isolation, and demonstrate a wide spectrum of potential presentations depending on neurologic structures compromised.
In contrast, a lower motor neuron lesion in which any component of the motor unit is disrupted (the motor neuron, the peripheral nerve, neuromuscular junction, or effector muscle fibers) would result in atrophied, flaccid musculature devoid of tone. If skeletal muscle fibers are denervated, the motor units may depolarize spontaneously, resulting in fasciculations. Patients complain of a weak, breathy, or diplophonic voice (the voice is heard at two distinct vibratory frequencies because each cord is at a different tension), and be unable to perform a glottal stop or valsalva maneuver. Endoscopic examination of the larynx would reveal impaired mobility of the vocal fold with possible bowing or flaccidity of the same fold and pooling of secretions in the ipsilateral pyriform sinus.
Lesions to the sensory pathways results in impaired gag, inability to clear secretions, and pooling of saliva. Patients may have gurgling speech, profound dysphagia, and absent or poorly productive cough.
The effects of lesions of the basal ganglia and cerebellum involve modulation of the frequency and smoothness of laryngeal movement. Due to the complex role of the basal ganglia, lesions involving its component structures present with laryngeal motion abnormalities that may be either hypokinetic or hyperkinetic. Hypokinetic disorders (parkinsonism) may yield a soft, breathy, monotonous voice, and difficulty initiating speech, with vocal fold bowing on phonation. Hyperkinetic disorders (e.g., dystonia, Huntington’s), may manifest with rapid unexpected vocal fold spasms with poor phonatory control. Disorders involving cerebellar pathways result in ataxic speech, with a disruption in fine control of volume and pitch, and a vocal tremor.
TABLE 70.1 NEUROLARYNGOLOGY EXAMINATION
Neurologic examination
•
Evaluate gait as patient walks to room
•
Complete head and neck examination
•
Cranial nerve examination:
○
Pay special attention to the tongue for tone and fasciculations: atrophy, flaccidity, and fasciculations indicate a LMN lesion, spasticity a UMN lesion
○
Look for twitching, dystonic posturing, or synkinesis of facial musculature
○
Pa, Pa, Pa: labial function
○
Ta, Ta, Ta/Ga, Ga, Ga: tip or posterior tongue function
○
Kitty cat: soft palate function
•
Look at outstretched hands for tremor
Vocal examination
•
Listen for fluidity (central), quality of articulation (central or peripheral), quality of the vocal signal (peripheral, laryngeal posturing, mucosal lesion)
Hyperfunction will give a strained quality that does not change significantly from one word to the next
○
Tremor will be rhythmic
○
Adductor spasms are best heard during voiced sounds, such as the /ay/ in 80
○
Abductor spasms can be elicited with a vowel following a voiceless consonant. In the word taxi, there will be involuntary breathiness during the /a/ sound after the voiceless consonant t.
Fiberoptic examination
•
Stop at level of the palate and evaluate for closure; have patient say “kitty cat”
•
With the patient breathing quietly through the nose, evaluate the glottis at rest for tremor or paradoxical motion
•
Evaluate for pooling of secretions
•
Evaluate glottal closure pattern, vocal fold motion, and supraglottic hyperfunction
•
Repeat vocal tasks during visualization and include
○
Glissando (sliding from low to high pitch): evaluates cricothyroid (CT) function
○
“Sniff: /i/” maneuver alternates adduction with abduction and will emphasize movement abnormalities such as a subtle paresis
○
Ask for purposeful inspiratory stridor, which may unveil lesions on the undersurface on the TVF
○
Agility and fatigability of vocal folds: alternate the sounds /pa/ – /ta/ – /ka/ past the point at which the patient is out of breath, assessing mobility, symmetry of motion, dexterity, and evidence of fatigue with repetition
•
At the end of the exam, gently touch the aryepiglottic folds with the tip of the endoscope to assess for laryngeal sensation
CT, cricothyroid; LMN, lower motor neuron; TVF, true vocal fold; UMN, upper motor neuron.
GENERAL APPROACH TO DIAGNOSIS AND TREATMENT
The diagnosis of neurologic disease of the larynx rests largely on the traditional history and physical examination (Table 70.1). Of particular importance are changes the patient has noticed in strength and movement outside of issues with phonation, as neurologic disease is rarely limited to just the larynx. A broad neurologic assessment and consultation with a neurologist may be warranted. Beyond the history and physical (which includes endoscopy), radiologic imaging, sensory testing and laryngeal electromyography (LEMG) can add additional information. Flexible transnasal laryngoscopy is preferable to transoral rigid laryngoscopy because the former alters laryngeal mechanics the least and allows the examination to be carried out across multiple laryngeal tasks.
Neurologic diseases can be classified on the basis of the neuroanatomical structures and systems affected. Not uncommonly, disorders may span a number of neuroanatomical structures, creating a mixed picture of dysfunction. The following categories focuses on disease entities most commonly associated with neurolaryngologic dysfunction, but is by no means exhaustive.
DISORDERS AFFECTING THE CENTRAL NERVOUS SYSTEM
Neurologic disorders of the central nervous system (CNS) are categorized on the basis of the systems affected. Lesions to the primary motor system (corticobulbar/corticospinal UMN, also known as pyramidal) primarily create spastic dysfunction, while lesions to the basal ganglia (extrapyramidal) result in disturbed control of function that may be hypo or hyperkinetic. (i.e., Parkinson disease would be hypokinetic, dystonia or tremor would be hyperkinetic). Disorders affecting the cerebellum and its outflow tracts in the brainstem and subcortical regions result in ataxic dysfunction. Some disorders are heterogeneous, resulting from inflammation or vascular occlusion to a number of important structures, with resulting combinations of dysfunction.
Spastic Disorders
Spastic disorders result from lesions or degeneration of UMN (pyramidal cells) and their projections in the corticobulbar pathway that extends from the cortex to the medulla. Stroke, the sudden onset of symptoms due to vascular compromise of cerebral tissue, commonly affects the motor pathways in addition to other structures and is discussed in the section under mixed CNS disorders. Amyotrophic lateral sclerosis (ALS), the most common nonvascular cause of spasticity, is a neurodegenerative disorder affecting both upper and lower motor neurons, and is discussed in detail under the section on peripheral nervous system disorders.
“Pseudobulbar palsy” refers to weakness of the bulbar musculature with hyperreflexia (hyperactive gag and brisk jaw jerk), indicating a UMN cause. This usually results from a lesion affecting the corticobulbar fibers. A true bulbar palsy would be due to compromise of lower motor neurons. Please see the explanation of bulbar palsy in the section on peripheral nervous system below.
Symptoms of pseudobulbar palsy include an exaggerated jaw jerk and gag reflex, and a spastic tongue that is contracted with a pointed tip but cannot be moved rapidly from side to side. Emotional lability (unprovoked crying or laughter), subcortical cognitive impairments, and dysarthria with thick, slurred, labored, and sometimes explosive articulation can be present. Marked hypernasality can exist, but palatal and pharyngeal reflexes are preserved. Dysphagia, dysarthria, nasal intonation, and nasal regurgitation are common.
Although often secondary to multiple bilateral vascular lesions above the brainstem, pseudobulbar palsy can also be caused by motor neuron disease. It is important to note that any disease involving the corticobulbar pathways can cause pseudobulbar palsy including multiple sclerosis (MS), neoplasm, encephalitis, and vascular disease.
Primary Lateral Sclerosis
Primary lateral sclerosis is a motor neuron disorder affecting UMN only and typically presents with spastic involvement of the lower extremities. There is often progression to spasticity of the bulbar musculature (2), giving the classic features of pseudobulbar palsy including spastic speech and dysphagia with pronounced emotional lability. The disorder is rare, accounting for a small percentage of all motor neuron disorders, and typically progresses very slowly (in contrast to ALS (3)). The cause is largely unknown. Differential diagnosis includes retroviral infection from HIV or human T-cell leukemia virus-I, MS, and adrenoleukomyelopathy. Antispasmodic pharmacotherapies such as baclofen (oral or intrathecal), or tizanidine can be helpful in ameliorating the spastic symptoms.
Hypokinetic (Parkinsonian) Disorders
Parkinsonism
Parkinsonism refers to the presentation of muscular rigidity and slowed movement (bradykinesia), with or without tremor. All forms involve damage to the pathways in the basal ganglia and brainstem, resulting in a net-inhibitory effect on movement and muscular function. Causes of secondary parkinsonism include stroke (usually accumulation of multiple basal ganglia lacunar infarcts), postencephalitic, paraneoplastic, traumatic (boxer’s encephalopathy), drug-induced (phenothiazines and other antipsychotic medications), and toxic (pesticides and designer drugs such as 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine, a meperidine analog) effects. Parkinsonism also includes a number of genetic and neurodegenerative disorders referred to as Parkinson Plus syndromes. These include Shy-Drager Syndrome (multiple systems atrophy or MSA), dementia with Lewy bodies, and progressive supranuclear palsy. Parkinson disease refers to idiopathic parkinsonism and is by far the most common hypokinetic movement disorder and also the most amenable to treatment. MSA is also relevant to otolaryngologists for its association with airway difficulties.
Parkinson Disease
Parkinson disease has a prevalence of 0.3% to 0.4% of the U.S. population (4) and affects up to 2% of people over age 60 across all ethnic groups with an equal sex distribution. Up to 80% demonstrate vocal difficulties, with 30% finding it the most disabling part of the disorder. The classic symptoms include a pill rolling tremor, cog-wheel rigidity, masklike facies, small handwriting, decreased blinking, flexed posture with shuffling gait, and reduced arm swing.
Pathologically, Parkinson disease is associated with the accumulation of inclusion granules of abnormal alphasynuclein protein in the autonomic and motor centers of the medulla oblongata. In later stages of disease, there are additional changes in the substantia nigra, basal forebrain, and ultimately, the frontal cortex (5). Autopsy examination shows loss of pigmentation of the substantia nigra, and cellular degeneration in the globus pallidus and putamen, with prominent Lewy body formation. Parkinson disease becomes symptomatic when the loss of substantia nigra dopaminergic neurons reaches about 70%. The cause for aberrant alpha-synuclein deposition in Parkinson disease is not known, but there are rare familial forms of the disease that include an autosomal dominant mutation in the alpha-synuclein gene (4q21) and an autosomal recessive mutation of the parkin gene (6q25.2-q27).
The vocal characteristics of Parkinson disease include a soft breathy monotone voice perceived by the patient to be of normal loudness. This is caused by poor effort from a bradykinetic bellows mechanism (chest wall and diaphragm) exacerbated by an inaccurate perception of personal speech effort. Additional components include vocal tremor, poor articulation, stuttering quality, difficulty initiating speech, and swallowing difficulties.
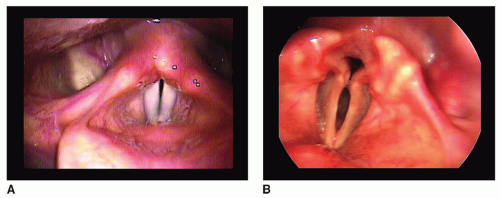
Laryngoscopy shows vocal fold bowing with a spindleshaped glottal gap on phonation (Fig. 70.3). Other findings include pooled hypopharyngeal secretions, decreased sensation, diminished cough reflex with aspiration, and poor coordination of the vocal folds with respiratory effort. Identification of vocal fold motion impairment suggests a parkinsonism plus syndrome, specifically MSA.
Pharmacotherapy for Parkinson disease aims to increase the amount of dopamine available in the synaptic cleft for neurotransmission to the basal ganglia. This can be accomplished by the use of the dopamine precursor levodopa (L-dopa, usually taken as an oral preparation with carbidopa, which inhibits peripheral metabolism of L-dopa), which is converted in the brain into dopamine by remaining substantia nigra cells. Other medications include dopamine agonists such as pramipexole and apomorphine (the latter available as an intravenous [IV] preparation), and therapies that prevent degradation and reuptake of dopamine once available in the synaptic cleft including the monoamine oxide type B inhibitor (selegiline) and catechol-O-methyltransferase inhibitors (entacapone). Thoughtful use of these medications is necessary, as spikes in dopamine levels can cause overflow movements including chorea and dyskinesias, while nadirs can result in ignition failure (inability to initiate movement) and freezing episodes.
Figure 70.3 Vocal fold bowing with Parkinson disease. A: Normal larynx in adduction, note the near complete closure of the membranous vocal folds and also the alignment of the vocal process of the arytenoid cartilages. B: This individual has the characteristic vocal fold bowing of parkinsonism —there is approximation of the tip of the vocal process of the arytenoids, but a prominent glottal gap at the midmembranous vocal fold and often (as seen here) a marked posterior glottal chink.
L-dopa has had mixed results in dysphonia, although most studies have shown no statistical improvement (6). Vocal fold augmentation or medialization laryngoplasty (7) may improve loudness and maximal phonation time if sufficient respiratory drive for phonation exists, but in general it has demonstrated only temporary effectiveness (most likely because of progression of disease). Laryngeal surgery does not improve articulatory function or prosody. Deep brain stimulation has shown variable improvement in speech, despite significant changes in limb movements (8,9). Only behavioral therapy has shown a consistent positive effect on voice and speech function of patients with Parkinson disease.
Lee Silverman Voice Therapy (LSVT) is the most well studied of the behavioral techniques for Parkinson disease (Table 70.2). The program works both by increasing patients’ physical efforts and also, theoretically, by resetting their sensory perception of their own vocal output. In the original study, 35 patients were randomized into placebo and treatment groups. At 2 years, the patients in the LSVT group sustained or improved vocal intensity over pretreatment levels, whereas the placebo group deteriorated (10). Swallowing function also appears to be improved after treatment (11).
Multiple Systems Atrophy
MSA combines parkinsonism and autonomic dysfunction. Patients with MSA suffer profound autonomic failure, usually manifesting as severe orthostatic hypotension, disturbance of sphincter control (urinary or fecal incontinence), and impotence. MSA-P (parkinsonian, previously “striatonigral degeneration”) has more resting tremor and parkinsonian features, and may be difficult to distinguish from Parkinson disease, save that these patients have a poor response to dopamine-related therapies. MSA-C (cerebellar, or olivopontocerebellar atrophy), has a greater degree of cerebellar ataxia. The prevalence is up to 5/100,000 persons (12). Like Parkinson disease, the pathogenetic mediator is the abnormal deposition of alpha-synuclein in intracellular inclusions (referred to as glial cytoplasmic inclusions, not Lewy bodies). Many patients have orofacial or craniocervical dystonia associated with a characteristic quivering, high-pitched dysarthria. Decreased sensation of the laryngopharynx and a diminished cough reflex may permit recurrent aspiration. Vocal fold immobility in a patient with parkinsonism is highly suggestive of MSA (13). Inspiratory stridor develops in about 30% of patients.
TABLE 70.2 LEE SILVERMAN VOICE THERAPY
Three major features contribute to the voice disorder in individuals with parkinson disease:
Global scale-down of neural drive to the speech mechanism, resulting in a soft monotone voice (bradykinesia)
Inaccurate sensory perception of self-effort involved in vocalization, which prevents the individual from accurately monitoring vocal output “My voice is not too soft, my spouse needs a hearing aid”
Difficulty with independently generating the correct effort to produce adequate vocalization “I can’t speak this loud, I feel I am shouting”
Five essential LSVT concepts:
Increasing amplitude of phonatory output (think loud, be loud)
Improving sensory perception of effort (think loud)
Administering treatment in high-effort style (recalibration)
Administering treatment intensively (four times a week for 16 sessions in 1 month)
Quantifying all speech and voice output (sound meter)
These individuals develop many sleep disorders, including obstructive and central sleep apnea, hypoxemia, phasic swings of systemic arterial pressure, reduced rapid eye movement (REM) sleep, and REM sleep behavior disorder. When obstructive sleep apnea occurs in patients with MSA, there is significant risk of sudden death (14), and tracheotomy is recommended. The disease progresses more rapidly than Parkinson disease, with average survival of less than 9 years.
Hyperkinetic Disorders
Dystonia
Dystonia is a chronic neurologic disorder of central motor processing within the basal ganglia (extrapyramidal system) characterized by involuntarily sustained muscle contractions of agonist and antagonist muscles causing twisting and repetitive movements or abnormal postures. Anxiety will aggravate the movements; sedatives may mitigate the movements; and posturing characteristically disappears during sleep. Dystonia can be focal (spasmodic dysphonia, writer’s cramp), regional (cervical dystonia), or generalized, and can be idiopathic or secondary to a wide variety of causes, including hereditary syndromes (Dystonia gene [DYT1] gene locus), stroke, trauma, and drug effects.
Spasmodic Dysphonia
Spasmodic dysphonia is a focal task-specific laryngeal dystonia with involuntary contractions of the intrinsic laryngeal musculature. If the contractions in the adductor musculature predominate, it is termed “adductor spasmodic dysphonia” (AddSD) and is characterized by a harsh, strained, and strangled quality with voice breaks. If the contractions in the abductor musculature predominate, it is termed abductor spasmodic dysphonia (ABdSD), which is characterized by sustained breathiness with breathy voice breaks. The dystonic (spasmodic) muscle contractions generally do not affect nonspeech laryngeal functions such as breathing, singing, swallowing, or coughing.
AddSD was first described by Traube in 1871 in a female having a “spastic form of nervous hoarseness” (spastiche form der nervosen helserkeit). Spasmodic dysphonia historically has been considered to have a psychogenic cause owing to the fluctuation in symptoms and frequent coexistence of anxiety and depression. However, in 1976, Dedo described the dramatic response (although temporary) of the disease to RLN section, providing proof of an organic cause (15). In many patients, the symptoms of anxiety and depression normalize after successful treatment of their spasmodic dysphonia (16). Although the exact pathophysiology of dystonia is unknown, some autopsy studies have detected altered levels of neurotransmitters in the caudate, putamen, globus pallidus, and dentate nuclei, supporting the current theory that the site of pathology involves the basal ganglia (17,18). Furthermore, recent studies suggest that dystonias involve cerebellar pathways that modulate extrapyramidal function (19).
Spasmodic dysphonia is an adult onset disorder with a female predominance. When classified, 83% of cases are AddSD, 17% ABdSD, with rare mixed cases and rare cases of breathing dystonia. Spasmodic dysphonia is associated with essential tremor in 30% of patients and with other forms of dystonia, such as blepharospasm or writer’s cramp, in 14% of patients (20). Up to 23% of patients with spasmodic dysphonia report a family history of dystonia or other movement disorders (21).
Spasmodic dysphonia affects the speaking voice but spares other vocal tasks such as singing, character speech (e.g., speaking in falsetto like Minnie Mouse), or yelling. Patients describe more difficulty with speaking over the telephone, public speaking, and communication during stressful situations. Alcohol or sedatives can reduce voice breaks, and speech during sleep is normal. Patients describe a “strangled” feeling when trying to speak, and have great difficulty “getting the words out.” Individuals with spasmodic dysphonia have a wide range of vocal patterns, depending on the severity of the spasms, compensatory vocal strategies used, and presence of overlying tremor. Some patients with AddSD use a low, breathy voice as a compensatory mechanism and sound similar to patients with ABdSD. Occasionally, it can be difficult to distinguish organic spasmodic dysphonia from psychogenic voice disorders or muscle tension dysphonia.
The vocal characteristics of AddSD include strangled breaks in connected speech, especially prominent with words that begin with vowels. Eliciting phrases would include counting from 80 to 90, and the sentence “We eat eggs every day.” The vocal characteristics of ABdSD include breathy breaks in connected speech, especially for vowels following a voiceless consonant. Eliciting phrases would include “The puppy bit the tape” and “Harry had a hard head.” The examiner should listen for delayed onset of phonation after the voiceless consonants p, t, and h.
Diagnosis is made by history, vocal characteristics, and flexible transnasal laryngeal examination across multiple speech tasks. Patients with AddSD demonstrate inappropriate adduction of vocal folds with hyperfunction of the supraglottis. Patients with ABdSD demonstrate inappropriate vocal fold abduction during connected speech. Some individuals are able to suppress symptoms with a sensory trick (geste antagonist) such as voicing while chewing, biting the tongue, or holding a finger in the corner of the mouth. Similarly, insertion of a flexible scope may diminish involuntary contractions and normalize speech. Associated neurologic disease should be excluded through neurologic consultation, preferably with a movement disorders specialist.
Botulinum toxin injection into the affected laryngeal muscles is the treatment of choice for the symptoms of spasmodic dysphonia, and results in vocal improvement in up to 90% of patients (21,22). For AddSD, the thyroarytenoidlateral cricoarytenoid muscle complex (TA-LCA) is most commonly injected; for ABdSD, the posterior cricoarytenoid (PCA) is injected. Complications are caused by excessive glottal weakness and breathiness with TA-LCA injections, stridor with PCA injections, and dysphagia to liquids if the toxin has diffused to the adjacent constrictors. Resistance to toxin at doses used to treat spasmodic dysphonia is rare, and most treatment failures are related to technical problems (improper dosing or placement of toxin).
With a successful injection, patients can expect reduction in voice breaks, effort required for speaking, and the uncomfortable strangling sensation. The adductor or abductor muscle group can be specifically targeted, and the dose titrated to provide just enough weakness to relieve spasm without causing excessive weakness and subsequent breathiness (Add) or stridor (ABd). Results are critically dependent on achieving sufficient weakness to ameliorate spasm without causing undue side effects.
Only gold members can continue reading. Log In or Register to continue