Neurogenic Tumors of the Orbit
Thomas C. Cannon
Keith D. Carter
Harry H. Brown
The orbit contains two types of nerves: the optic nerve (a tract of the central nervous system) and peripheral nerves (both branches of sensory nerves, such as the fifth nerve, and motor nerves, such as those that innervate the extraocular muscles). This chapter is concerned primarily with tumors of neurogenic origin that have arisen from peripheral nerves. The complex diversity of tissues within the orbit is surpassed by no other anatomic site. Primary orbital tumors that are neurogenic range from relatively common entities, such as the benign peripheral nerve sheath tumors, neurofibroma and schwannoma, to the extremely rare orbital tumors, such as malignant peripheral nerve sheath tumor, paraganglioma, and primitive neuroectodermal tumor.
To better understand the composition of neurogenic tumors of the orbit, we first must understand the basic histologic composition of a peripheral nerve. A peripheral nerve is composed of several bundles of axons referred to as fascicles. Within the peripheral nervous system, an axon is an extension of an individual nerve cell body. Each axon is invested by Schwann cells that provide the myelination for each individual axon of a fascicle. Between the individual axons is the endoneurium. The endoneurium is composed of amorphous extracellular matrix material and collagen fibrils, which provide the infrastructure for the support of the axons. The perineurium is the connective tissue that surrounds each fascicle (Fig. 41-1). The epineurium encompasses the entire nerve and contains the blood supply of the nerve.
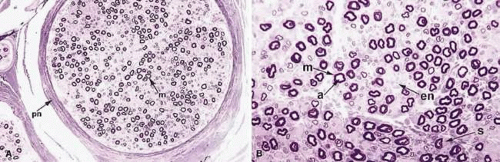 Fig. 41-1. Histologic cross sections of a peripheral nerve. m, myelinated fiber; pn, perineurium; a, axon; en, endoneurium; Schwann cell(s). (Toluidine blue; A, 75X; B, 300X.) |
NEUROFIBROMAS
Neurofibromas are benign peripheral nerve sheath tumors composed of axons, Schwann cells, and fibroblasts, in contrast to schwannomas, which are composed predominantly of Schwann cells. Neurofibromas represent from 0.5% to 2.4% of all orbital tumors.1 Neurofibromas may be classified into three major categories: isolated (localized) neurofibroma, diffuse neurofibroma, and plexiform neurofibroma. Some authors also categorize the traumatic neuroma as a fourth category of neurofibroma, although it is a reactive proliferation.2
ISOLATED NEUROFIBROMAS
Isolated (localized) neurofibroma is so named because it generally grows as a circumscribed mass. Although circumscribed, it usually is not encapsulated, as is the schwannoma. Isolated cutaneous neurofibromas may develop anywhere on the body and rarely produce symptoms. Isolated orbital neurofibromas tend to involve the superior aspect of the orbit but manifest in the third to fifth decades (Fig. 41-2A). Decreased visual acuity is an uncommon symptom of localized neurofibroma confined to the anterior compartment of the orbit. When the posterior aspect of the orbit is affected, compression of the optic nerve may lead to decreased visual acuity and visual field defects and may produce a relative afferent pupillary defect. Diplopia, which also may develop in isolated neurofibromas of the posterior aspect of the orbit, more likely is the result of mass effect rather than direct involvement of the nerves innervating the extraocular muscles.
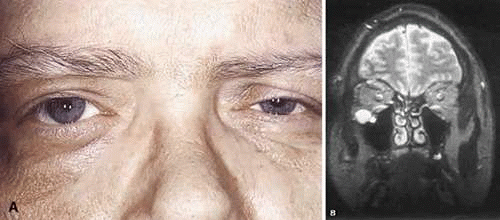 Fig. 41-2. A: Isolated (localized) orbital neurofibroma with mild proptosis of the right eye. B: On MRI, the lesion appears to be circumscribed in the inferior aspect of the orbit. Despite the circumscribed radiologic appearance, these tumors are not encapsulated. |
Radiographic studies often demonstrate the circumscribed nature of the tumor, although isolated neurofibromas are not encapsulated like schwannomas (Fig. 41-2B). Some authors consider the classification of a neurofibroma as localized neurofibroma only in the absence of documented neurofibromatosis. Multiple localized orbital neurofibromas have been reported in patients who do not have neurofibromatosis.3,4 It is unclear whether these cases actually represent a forme fruste of neurofibromatosis.
Histologically, localized neurofibroma is characterized by wavy spindle cells with eosinophilic cytoplasm and comma-shaped nuclei (Fig. 41-3). Collagen is present in variable quantities. Nuclear palisading is not typical, in contrast to schwannoma. Ultrastructurally, neurofibroma is composed predominantly of cells that have features of the perineurial fibroblasts. In contrast, schwannomas have cellular homogeneity, extracellular long-spacing collagen, and abundant basement membrane.
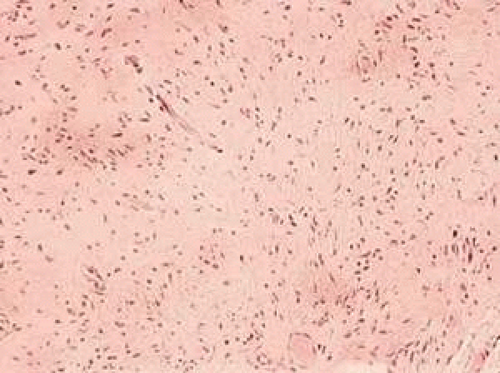 Fig. 41-3. Localized neurofibroma. Slender spindle cells with characteristic wavy nuclei are enmeshed in a fibrillar stroma. (Hematoxylin-eosin, 75X.) |
Treatment of localized neurofibroma depends on whether the mass is producing symptoms, and on its location. Within the orbit most are well delineated by imaging studies such as computed tomography (CT) or magnetic resonance imaging (MRI). Surgical excision through an anterior orbitotomy usually is curative.
DIFFUSE AND PLEXIFORM NEUROFIBROMAS
Diffuse neurofibroma and plexiform neurofibroma both are associated with neurofibromatosis; plexiform neurofibroma is considered pathognomonic of neurofibromatosis type 1. Both types of tumors tend to present within the first few decades of life. Diffuse neurofibroma is not circumscribed. Many diffuse neurofibromas are solid, although they may have a grossly myxoid or gelatinous appearance at the time of surgery. Both grossly and microscopically diffuse neurofibromas feature an infiltrative growth pattern, with tumor cells separating and/or infiltrating adipose tissue on histologic examination (Fig. 41-4). Likewise, the radiographic appearance is infiltrative compared with the circumscribed nature of the localized neurofibroma and schwannoma. The plexiform neurofibroma also lacks encapsulation, and radiographic evaluation may not distinguish plexiform from diffuse neurofibroma.
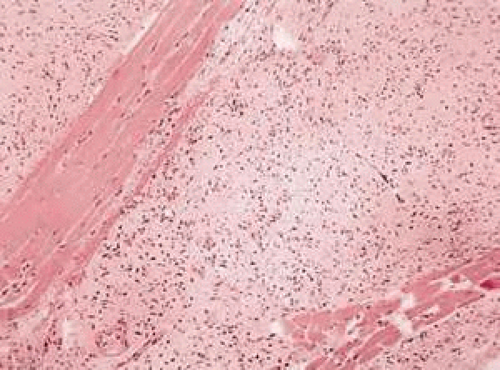 Fig. 41-4. Diffuse neurofibroma with the characteristic wavy nuclei, as in Figure 41-3. This tumor dissects between fascicles of skeletal muscle. (Hematoxylin-eosin, 75X.) |
Plexiform neurofibroma is the most common orbital manifestation of neurofibromatosis type 1. Patients with neurofibromatosis have a greater likelihood of developing a malignant peripheral nerve sheath tumor than individuals in the general population. Plexiform neurofibromas most commonly develop within large peripheral nerves. These tumors tend to distort multiple segments of large nerves, most commonly in the extremities. This diffuse thickening experienced on palpation of these lesions has been referred to as a “bag of worms” (Fig. 41-5). Elephantiasis neuromatosa occurs when diffuse thickening of the nerve leads to enlargement and distortion of an entire extremity. A similar orbital presentation may be caused when plexiform neurofibroma involves the eyelids and surrounding facial nerves and tissues.
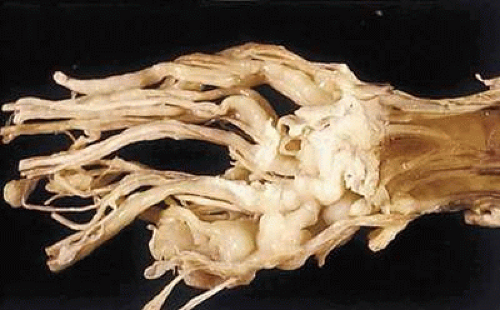 Fig. 41-5. Gross photograph of a plexiform neurofibroma of the cauda equina taken at postmortem examination. The patient died of malignant peripheral nerve sheath tumor (not illustrated). Plexiform neurofibromas and malignant peripheral nerve sheath tumors may involve the orbit. |
Histologically, plexiform neurofibromas have several unique characteristics. Microscopically, they are composed of aggregates of tortuous branches of expanded nerves that appear disorganized when cut in different planes of section. These tumors may have an increased amount of myxoid extracellular matrix material (Fig. 41-6). Special stains, such as silver stains, can be used to identify axons randomly distributed within these lesions, as opposed to schwannomas, which displace the axons to the periphery of the mass. Schwannomas, often invade the nerves, and entrapped axons can be identified with these staining methods.
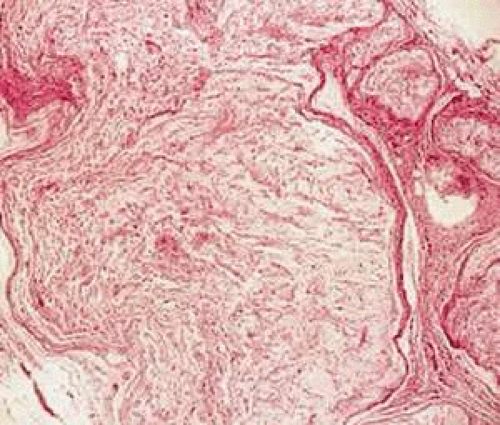 Fig. 41-6. Histologic section of a plexiform neurofibroma with disorganization and expansion of the nerve fibers and extracellular myxoid matrix. (Hematoxylin-eosin, 150X.) |
The major differentiating feature between the diffuse neurofibroma and the plexiform neurofibroma, however, is the clinical appearance. The plexiform neurofibroma causes a diffuse enlargement and distortion of the nerve and a characteristic eyelid deformity (Fig. 41-7).
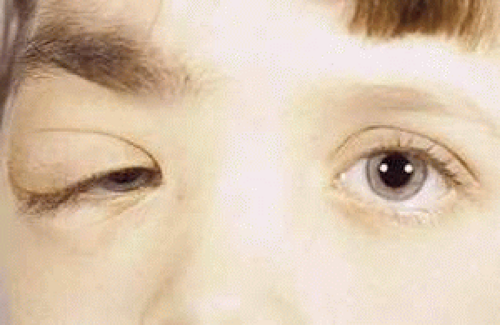 Fig. 41-7. Child with neurofibromatosis type 1 and orbital plexiform neurofibroma with extensive involvement of the eyelid. |
SCHWANNOMAS
Schwannomas (neurilemomas) are benign peripheral nerve sheath tumors that may arise either de novo or as a manifestation of neurofibromatosis. Schwannomas in the orbit most frequently develop from the superior ciliary nerves, although they also may arise from smaller peripheral nerves. Orbital schwannomas are saccular enlargements of the involved nerve and appear to be encapsulated enlargements of the involved nerve and therefore displace the nerve to one side.
Clinically, orbital schwannomas represent approximately 1% of all orbital tumors and are found in 1.5% of all patients with neurofibromatosis type 1. They typically occur in young to middle-aged people and present as slowly progressive proptosis (Fig. 41-8A) that may have evolved over years or decades. Presenting complaints vary according to location of the tumor within the orbit. Tumors at the orbital apex may present earlier because their compressive effects are more immediately symptomatic. In a literature review of symptomatic orbital schwannomas by Cantore et al.,5 in 64 patients, 43 patients had exophthalmos and 49 patients had impairment of ocular motility, while deficits in visual acuity were seen in 28 patients, and optic disc abnormalities also were seen in only 28 patients (Fig. 41-8B-D).
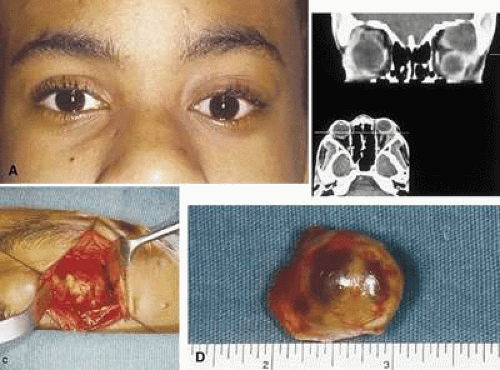 Fig. 41-8. A: Orbital schwannoma in a 16-year-old boy showing proptosis. B: CT scan of the orbit reveals an encapsulated schwannoma in the superior temporal aspect of the right orbit. The left eye is displaced downward (globe ptosis), and the eye does not appear to be the same size on this cut as the right eye. The discrepancy in the apparent size of the eye in this imaging plane is accounted for by the proptosis on the left side. C: Intraoperative photograph shows that the tumor is encapsulated. D: Surgically excised orbital schwannoma. A glistening capsule wraps around the tumor. The two dark red areas beneath the capsule represent zones of focal cystic degeneration. |
Schwannomas often have distinctive histologic features that can facilitate their pathologic diagnosis. The main histologic components that are helpful, when present, include Antoni A and Antoni B type areas, and Verocay bodies. Antoni A refers to a hypercellular histologic pattern of the tumor, while Antoni B is textured more loosely, with cystic spaces present. The Verocay body is the name given to the palisading of the nuclei of schwannomas associated with acellular zones (Fig. 41-9). Verocay bodies, when present, are a useful marker for schwannoma. Less well developed palisading of the cell nuclei is a frequent finding (Fig. 41-10). Other histologic features include hyalinization of vessel walls, hemosiderin-laden histiocytes, and foamy histiocytes. Encapsulation is due to saccular expansion of the involved nerve with preservation of the perineurium and is manifest histologically by a band of eosinophilic fibrous tissue surrounding the tumor (Fig. 41-11). When the histopathologic examination cannot clearly differentiate between schwannoma and other tumors, such as fibrous meningioma, immunohistochemistry and electron microscopy can be performed. Immunohistochemical stains for S-100 frequently are positive in schwannoma and not in meningioma. By electron microscopy, Schwann cells have abundant basement membrane and long-spacing collagen (Luse bodies).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


