Essentials of Diagnosis
General Considerations
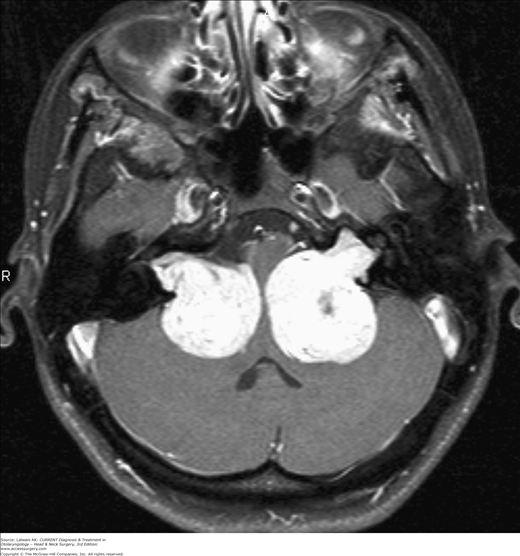
Neurofibromatosis type 2 (NF2) is the official name for the syndrome whose hallmark is bilateral vestibular schwannomas (VS) (Figure 63–1). NF2 replaces a variety of synonyms that have been associated with this entity: central neurofibromatosis, bilateral acoustic neurofibromatosis, cranial neuromatosis, central schwannomatosis, neurofibromatosis universalis, familial bilateral acoustic neuroma syndrome, familial bilateral acoustic neurofibromas, Wishart–Gardner–Eldridge syndrome, neurinomatosis, and neurofibrosarcomatosis. The first known description of the clinical course and postmortem findings of NF2, almost two centuries ago, was of a patient who developed bilateral deafness, had intractable headaches and vomiting, and died at age 21.
NF2 has long been confused with classic von Recklinghausen syndrome and has only recently been recognized as a distinct diagnostic entity. In 1987, a consensus panel of the National Institutes of Health officially differentiated the clinical manifestations associated with classic von Recklinghausen syndrome or peripheral neurofibromatosis from those of a predominantly intracranial subtype or central neurofibromatosis. The two syndromes were designated NF1 and NF2, respectively. Molecular genetic investigations confirmed this clinical differentiation: the gene responsible for NF1 was located near the proximal long arm of chromosome 17, whereas the gene responsible for NF2 was located on chromosome 22.
NF2 is much rarer than NF1, with an incidence estimated between 1:33,000 and 1:50,000. Inheritance of NF2 is autosomal dominant and gene penetrance is above 95%. NF2 most frequently presents in the second and third decades of life. VS represent approximately 8% of intracranial tumors and account for approximately 80% of the tumors found in the cerebellopontine angle. Most cases of VS occur sporadically, are unilateral, and present in the fifth decade. Patients with NF2 have bilateral VS and represent 2–4% of patients with VS.
The recent identification of the gene responsible for NF2 has significantly advanced our understanding of the molecular pathology, as well as the factors responsible for the clinical heterogeneity among patients with NF2. The NF2 gene encodes for the protein merlin or schwannomin, has been shown to have homology to the ezrin–radixin–moesin family of genes, which functions as membrane-organizing proteins. These proteins have a basic function indigent to all cells, which are postulated to link cytoskeletal proteins to the plasma membrane. It has been proposed to represent a recessive tumor suppressor, whose deletion or inactivation alters the abundance, localization, and turnover of cell-surface receptors, thus initiating tumorigenesis. Understanding the function of merlin in tumor formation will lead to the development of novel therapies that may eventually alleviate the suffering associated with NF2.
Pathogenesis
NF2 results from the inheritance of a mutation in merlin (or schwannomin) protein on chromosome 22. The NF2 gene is spread over approximately 100 kb on chromosome 22q12.2 and contains 17 exons. The coding sequence of the messenger RNA is 1785 bp in length and encodes a protein of 595 amino acids. The gene product is similar in sequence to a family of proteins the include moesin, ezrin, radixin, talin, and members of the protein 4.1 superfamily. These proteins are involved in linking cytoskeletal components with the plasma membrane and are located in actin-rich surface projections such as microvilli. The N-terminal region of the merlin protein is thought to interact with components of the plasma membrane and the C-terminal with the cytoskeleton. Although the exact function of the NF2 protein is as yet unknown, the evidence available so far suggests that it is involved in cell–cell or cell–matrix interactions and that it is important for cell movement, cell shape, and communication. The loss of function of the merlin protein therefore could result in a loss of contact inhibition and consequently lead to tumorigenesis. NF2 gene defects have been detected in other malignant disorders including meningiomas, malignant mesotheliomas, melanomas, and breast carcinomas.
Approximately 50% of affected patients have no family history of NF2. Therefore, these patients represent new germ line mutations in the NF2 gene. To date, more than 200 mutations of the NF2 gene have been identified, including single-base substitutions, insertions, and deletions. Genotype–phenotype correlation studies suggest that mutations in the NF2 gene, which result in protein truncation, are associated with a more severe clinical presentation of NF2 (Wishart type), whereas missense and splice site mutations are associated with a milder (Gardner type) form of the disease. Retinal abnormalities were associated with the more disruptive protein truncation mutations of the NF2 gene. Although mutations in the NF2 gene play a dominant role in the biology of VS, it is also possible that other genetic loci contribute to the development of VS.
Clinical Findings
Patients with NF2 usually present in the second and third decades of life, rarely after age 60. Patients’ symptoms are attributable to VS, cranial meningiomas, and spinal tumors. The presentation of NF2 can vary considerably, but has been broadly divided into two subtypes based on the severity of disease: Gardner and Wishart NF2 subtypes (Table 63–1). The more severe Wishart type of NF2 is characterized by an early onset of tumors, a more rapid course of disease progression, and the presence of multiple other tumors in addition to bilateral VS. In contrast, the milder Gardner subtype is characterized by a later onset of symptoms, a more benign course of disease, and a tumor burden usually limited to bilateral VS. Many patients with NF2, however, cannot easily be categorized into these subtypes and have many overlapping features.
| Gardner | Wishart |
|---|---|
|
|
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


