Chapter 57 Neurofibromatosis 2
NEUROFIBROMATOSIS 2 DIFFERENTIATED FROM NEUROFIBROMATOSIS 1
CLINICAL CHARACTERISTICS OF NEUROFIBROMATOSIS 2
Definition
The NIH Consensus Development Conference also developed guidelines for the diagnosis of NF2. NF2 is distinguished by bilateral vestibular schwannomas with multiple meningiomas, cranial tumors, optic gliomas, and spinal tumors. A definite diagnosis is made on the basis of the presence of bilateral vestibular schwannomas or developing a unilateral vestibular schwannoma by age 30 and a first-degree blood relative with NF2, or developing at least two of the following conditions known to be associated with NF2: meningioma, glioma, schwannoma, or juvenile posterior subcapsular lenticular opacity/juvenile cortical cataract (Table 57-1).1
TABLE 57-1 Neurofibromatosis 2 (NF2) Diagnostic Criteria
| INDIVIDUALS WITH THE FOLLOWING CLINICAL FEATURES HAVE CONFIRMED (DEFINITE) NF2 |
| Bilateral VS or family history of NF2 (first-degree family relative) plus |
| Unilateral VS <30 years or |
| Any two of the following: meningioma, glioma, schwannoma, juvenile posterior subcapsular lenticular opacities/juvenile cortical cataract |
| INDIVIDUALS WITH THE FOLLOWING CLINICAL FEATURES SHOULD BE EVALUATED FOR NF2 (PRESUMPTIVE OR PROBABLE NF2) |
| Unilateral VS <30 years plus at least one of the following: meningioma, glioma, schwannoma, juvenile posterior subcapsular lenticular opacities/juvenile cortical cataract |
| Multiple meningiomas (≥2) plus unilateral VS <30 years or one of the following: glioma, schwannoma, juvenile posterior subcapsular lenticular opacities/juvenile cortical cataract |
VS, vestibular schwannomas.
There may be significant heterogeneity in the presentation of the disease from one individual to the next. Some individuals may have a very mild form of the disease with small vestibular schwannomas manifesting in an older individual. Meanwhile, some children present with multiple intracranial tumors at a very young age. Despite the heterogeneity of the disease within a family, the expression of NF2 tends to be very similar.2 There is a significant genetic component to the disease with much variability within the parameters of the observed phenotype. Studies have shown that a truncating mutation (nonsense and frame shift) may be linked with a more severe form of NF2.3–5 The more severe form of NF2 is termed Wishart form. Individuals with this severe form present with early onset of the disease with multiple intracranial schwannomas and meningiomas that result in blindness, deafness, paralysis, and death by age 40. Despite the strong genotype-phenotype correlation, individual differences in tumor growth occur within subjects, making it difficult to predict how an individual will change over time even when the genotype is known.
The milder form, or Gardner form, of NF2 is less debilitating. The schwannomas may remain stable for many years, few meningiomas develop, and patients may not develop symptoms until later in life and often have fewer disabilities. The genetic basis of the mild form has not been well characterized. Many of these may be mosaic forms of the disease, however.2-4,6-12
Prevalence and Incidence
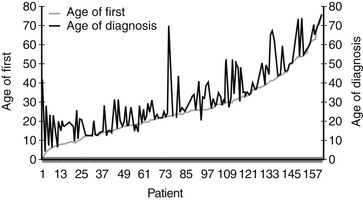
The average age of diagnosis of NF2 is 25 years; however, many patients present with symptoms before the diagnosis. There is an average delay of diagnosis of approximately 7 years (Fig. 57-1). There is no difference in the proportion of men versus women who develop NF2, and no prevalence has been described based on ethnicity. Epidemiologic studies place the incidence of NF2 between 1 in 40,000 live births13 and 1 in 87,410 live births.14
Molecular Genetics
The NF2 gene was mapped to chromosome 22 q 12-2in 1993.15–17 The NF2 gene located at chromosome 22 codes for a tumor suppressive protein termed Merlin or Schwannomin. This protein negatively regulates Schwann cell production. The loss of this protein allows overproduction of Schwann cells. The mutation in the NF2 chain predisposes individuals to developing a schwannoma when the second hit occurs to the gene; control of Schwann cells is lost or mutated within the cell. Various types of mutations have been identified, including single base substitutions, insertions, and deletions.4,18–20 The mild, or Gardner, type of NF2 may be associated with missense mutations, whereas associations between the other mutations and phenotypes are not as clear.21 The occurrence of NF2 is not restricted to families known to carry the mutation. Frequently, genetic mosaicism occurs, which may not be detected by common mutation analysis techniques.22 Unilateral vestibular schwannomas may exhibit the same type of genetic markers as NF2.23 The mutations in unilateral vestibular schwannomas are confined to the affected tumor tissue. In patients with NF2, the mutation is present in all cell types.22
Tumor Types
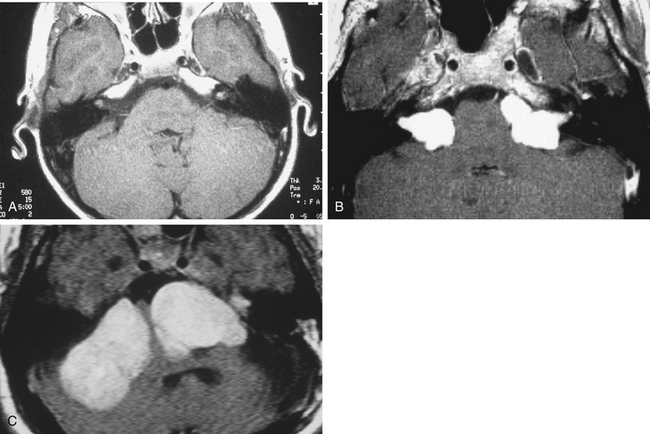
Bilateral vestibular schwannomas (acoustic neuromas) are benign neoplasms of the acoustic or eighth cranial nerve (Fig. 57-2).24 The tumors are thought to arise at the glioma–Schwann’s cell junction within the internal auditory meatus. The tumors most commonly arise from the superior vestibular nerve, although with NF2, tumors may be found on the cochlear and facial nerves within the internal auditory meatus. The consequences of a vestibular schwannoma are numerous, including hearing loss progressing to deafness, dizziness and balance problems, tinnitus, facial nerve paralysis, brainstem compression, and, if left untreated, death.
Despite the strong genetic effect in NF2, there is enormous variability in the number of tumor types, the rate of progression, and the disabilities experienced. This enormous variability is also found in patient presentation. Some patients may be asymptomatic. Patients who have no symptoms when diagnosed have generally been identified on the basis of genetic analysis conducted because a blood relative has NF2 or presymptomatic screening. Although the NIH criteria for NF2 require the presence of bilateral vestibular schwannomas for diagnosis, patients may first develop unilateral schwannomas as a young child with no other tumors, or adult patients may present with multiple meningiomas (cranial and spinal) and no vestibular schwannomas.9,25 Although the NIH criteria for NF2 imply that all NF2 patients develop bilateral vestibular schwannomas, some researchers are not convinced of this.36 Evans and colleagues26 based their conclusion on the observation of a possible variant form of NF2 manifesting with skin and spinal tumors in the absence of vestibular schwannomas. Nonetheless, the phenotype generally is reflective of the underlying disorder.
Intracranial Schwannomas
Vestibular schwannomas are the most common intracranial schwannoma associated with NF2. The most frequently identified nonvestibular schwannomas are schwannomas of CN III and V. Bilateral CN III or V schwannomas are the most common additional schwannomas seen. It is important to identify these lesions on MRI. Lower cranial nerve schwannomas may also be identified, but are much less frequently seen. A vestibular schwannoma rarely turns malignant, and sometimes the unilateral vestibular schwannoma may regress in size altogether. Growth of the tumors does not seem to be related either to loss of heterozygosity (genetic level of analysis) or to auditory functioning (phenotype level of analysis). For this reason, it is recommended that a patient have at least yearly MRI scans to track changes in size.33–40 All newly diagnosed patients should have a full head and spine study to stage their disease. After the disease is staged, a 6 month study is performed to determine if the tumor is fast-growing or slow-growing.41
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


