Neonatal Respiratory Distress
Mark Boston
Respiratory distress occurs in up to 8% of newborn infants and may be manifested by tachypnea, increased work of breathing, nasal flaring, use of accessory muscles of respiration, inspiratory retractions, and cyanosis. Cardiopulmonary problems are the most frequent reasons for neonatal respiratory distress. Sepsis, hypoglycemia, hematologic and metabolic abnormalities, central hypoventilation, and airway obstruction may also cause respiratory distress in the neonate (1).
Congenital upper airway anomalies are rare but can cause significant respiratory distress during the neonatal period (2). Dynamic or fixed obstruction anywhere from the nose and mouth to the tracheobronchial tree may result in respiratory distress. The signs and symptoms of neonatal airway obstruction include noisy breathing (stridor or stertor), feeding difficulties, sternal and intercostal retractions, tachypnea, hypercarbia, hypoxia, cyanosis and, in severe cases, apnea, bradycardia, and cardiac arrest. While the signs and symptoms are often characteristic of the site of obstruction—nasal, oropharyngeal, laryngeal or tracheobronchial—significant overlap exists among the anatomic sites and their presenting symptoms. Therefore, a thorough evaluation of the distressed neonate is essential and includes a detailed history and physical and directed endoscopic, radiographic, and laboratory studies.
ANATOMY AND PHYSIOLOGY
The unique nature of the infant airway must be appreciated in order to appropriately evaluate and manage neonatal respiratory distress. The neonatal larynx is situated high in the neck between the second and fourth cervical vertebrae and forms an obtuse angle with the mandible. The high position of the larynx brings the tip of the epiglottis into contact with the soft palate, making the neonate an obligate nasal breather. The neonatal larynx is softer, collapses easily with higher negative airway pressures, is easily irritated, and is more prone to laryngospasm than is the adult larynx. Laryngeal spasm may result in bronchial constriction, apnea, and bradycardia with life-threatening consequences. Neonates also have small-caliber airways with high resistance, low lung compliance, high chest wall compliance, low oxygen reserve, and high oxygen consumption. As a result, neonates do not tolerate diminished ventilation or apnea for more than a few seconds without developing significant hypoxemia. Therefore, during endoscopic evaluation of the neonatal airway, diligence is required with respect to the appropriate depth of anesthesia, topical anesthesia of the larynx, gastric distention, and blood oxygen saturation monitoring.
EVALUATION
When presented with a neonate in respiratory distress, the physician must first decide if the airway needs to be emergently managed or if he or she can proceed with a detailed history and physical exam (Fig. 88.1). The physician should be able to determine if urgent airway intervention is required within a few seconds by carefully observing the child’s level of consciousness, coloring, respiratory effort and listening for airway noise. With a stable or stabilized airway, the assessment may continue in order to determine the specific etiology of the airway obstruction. In all cases of neonatal airway distress, the history must include a detailed accounting of the pre- and perinatal histories, family history, onset and timing of airway noise, associated feeding difficulties, and the presence or absence of other associated signs and symptoms. Examination may follow the history or proceed simultaneously with active airway management in the severely distressed neonate.
The history, time of onset, and identification of initiating factors are helpful in the search for the cause of stridor in the neonate. The effects of positioning, feeding, or crying offer further diagnostic clues. A history of prematurity, difficult
labor or delivery, or intubation may be contributory. The quality of stridor and its relationship to the respiratory cycle offer valuable diagnostic clues as does the character of the newborn’s cry and the presence of dysphagia or a cough. The neck and lungs should be auscultated and, after completing the history and general physical examination, the airway should be examined endoscopically.
labor or delivery, or intubation may be contributory. The quality of stridor and its relationship to the respiratory cycle offer valuable diagnostic clues as does the character of the newborn’s cry and the presence of dysphagia or a cough. The neck and lungs should be auscultated and, after completing the history and general physical examination, the airway should be examined endoscopically.
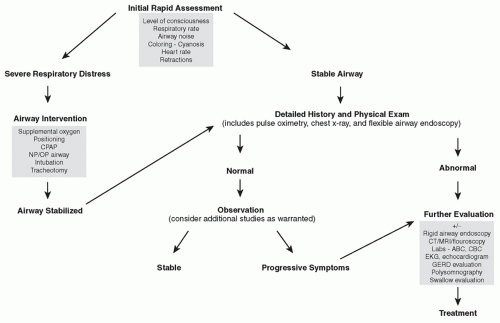 Figure 88.1 Algorithm for the evaluation and treatment of neonatal airway obstruction. |
Flexible endoscopy using a pediatric flexible laryngoscope should be performed in all neonates with suspected airway obstruction. Care must taken, however, to ensure that the patient is stable for the exam and that the procedure will not exacerbate the airway problem. The pediatric flexible laryngoscope is used to assess the nose, nasopharynx, oropharynx, and larynx to the glottis and the upper hypopharynx. However, the pyriform sinuses, posterior cricoid region, and subglottis cannot be consistently examined with this technique. Whenever possible, the study should be performed with a camera attachment and recorded for documentation and review. Application of a topical nasal decongestant may be helpful, but the routine use of topical anesthesia is not necessary (3).
Not all neonates with suspected congenital airway obstruction need undergo rigid endoscopic examination under anesthesia if the diagnosis can be obtained confidently by a combination of flexible endoscopy and/or radiographic studies; however, if patients show a progression of symptoms marked by increasing respiratory distress or failure to thrive, microlaryngoscopy, bronchoscopy, and esophagoscopy are indicated for both diagnostic and therapeutic reasons.
Continuous pulse oximetry monitoring and a chest radiograph are standard for all neonates in respiratory distress (1). Additional studies are directed based on the history, general physical examination, endoscopic examination, and radiographic findings. In patients with suspected tracheomalacia, videofluoroscopy helps delineate the full extent of luminal collapse and diaphragmatic excursion (4). A contrast esophagogram can be useful in the diagnosis of tracheoesophageal fistulae, esophageal stenosis, and vascular anomalies or rings that cause extrinsic compression. Detailed information regarding airway stenosis, mass lesions, or thoracic vascular abnormalities may be provided by computed tomography (CT) or magnetic resonance imaging (MRI) (5). An echocardiogram and electrocardiogram are useful to assess for cardiac abnormalities and possible cardiac strain. An evaluation for gastroesophageal reflux disease (GERD) will be appropriate in selected patients and may include a pH probe study, esophagoscopy with biopsies, barium swallow, or esophageal scintiscan. When aspiration is suspected, a functional endoscopic evaluation of swallowing (FEES) may be useful.
After the diagnosis has been obtained, a treatment plan is instituted. In the patient with progressive respiratory distress but no identified airway obstruction, the physician should consider pulmonary, cardiac, central nervous, gastrointestinal, iatrogenic, or metabolic-nutritional causes of respiratory failure. If there is an upper airway abnormality, bypassing the obstruction should relieve the symptoms of respiratory distress. With coexisting pulmonary pathology, however, the patient may remain symptomatic with tachypnea, hypoxemia, and hypercarbia, and require ventilatory assistance. When upper airway pathology has been eliminated as a cause of the patient’s respiratory distress, the neonatologist should assume primary management of the patient.
NOSE
Because infants are initially obligate nasal breathers, nasal obstruction frequently causes severe respiratory distress in neonates. Nasal obstruction typically results in stertor, a sonorous or snorting type of respiration, with nasal flaring. With complete nasal airway obstruction (e.g., bilateral choanal atresia) cyanotic episodes are frequent and may be alleviated during crying, a phenomenon known as cyclical cyanosis. Chest retractions are common and feeding is difficult because patients must breathe and feed through their mouths at the same time. Severe respiratory distress due to nasal obstruction should be managed with an oral airway or intubation prior to completing the history and examination.
Examination begins with an external assessment of the nose and surrounding facial structures. Small pits or dimples along the midline of the nose suggest a nasal dermoid (6). These may be found from the nasal dorsum to the columella. Similarly, a midline nasal mass may represent a cephalocele, glioma, teratoma, or extension of an intracranial vascular anomaly. Craniofacial anomalies, including Treacher Collins, Crouzon, and Apert syndromes, may present with nasal obstruction, and recognition of these anomalies should direct the examination to the nose and nasopharynx (7). Large intranasal hemangiomas or vascular malformations may also cause respiratory distress and may cause distortion of the external nose.
After the external assessment, a thorough intranasal examination should be undertaken. The most common cause of neonatal nasal airway obstruction is neonatal rhinitis. The patient with neonatal rhinitis typically presents with stertor and feeding difficulties. Examination reveals diffuse nasal mucosal edema and turbinate hypertrophy with a normal pyriform aperture and patent choanae. Treatment includes the use of nasal saline drops and or 0.1% dexamethasone ophthalmic drops into both nostrils for several days. GERD may also contribute to neonatal rhinitis and should be treated if suspected based on the history and physical examination.
Anterior nasal stenosis is uncommon, but obstruction at the level of the pyriform aperture secondary to medial overgrowth of the maxilla may cause distress. Airway distress may be especially prominent if there is associated turbinate hypertrophy. Examination of the intranasal structures with a flexible nasopharyngoscope or rigid telescope can identify the cause of obstruction. Inability to pass an endoscope through the anterior nose due to narrowing of the nasal fossa should alert the clinician to the possibility of pyriform aperture stenosis. Although rare, pyriform aperture stenosis can be associated with a single upper central incisor or other craniofacial dysmorphology (8,9). The inferior meatus should be examined carefully for nasolacrimal duct cysts (Fig. 88.2). Passing the nasopharyngoscope more posteriorly allows examination of the posterior choanae and nasopharynx. If choanal atresia is suspected because of the inability to pass either a nasopharyngoscope or a small feeding tube more than 5.5 cm from the alar rim, CT evaluation is appropriate (Fig. 88.3) (10,11). Children with either pyriform aperture stenosis or choanal atresia should be assessed by a geneticist because of a high incidence of concomitant craniofacial and developmental anomalies (e.g., CHARGE syndrome) (10). Adenoid hypertrophy or a nasopharyngeal tumor also may be identified as a cause of nasal airway obstruction.
Radiographic assessment of neonates with nasal obstruction is often necessary, and CT is the preferred study for most cases of neonatal nasal obstruction. In some conditions, however, including midline nasal masses with potential intracranial connections, MRI may also be indicated (6). Consultation with a pediatric neuroradiologist is recommended to optimize the radiographic evaluation of these patients.
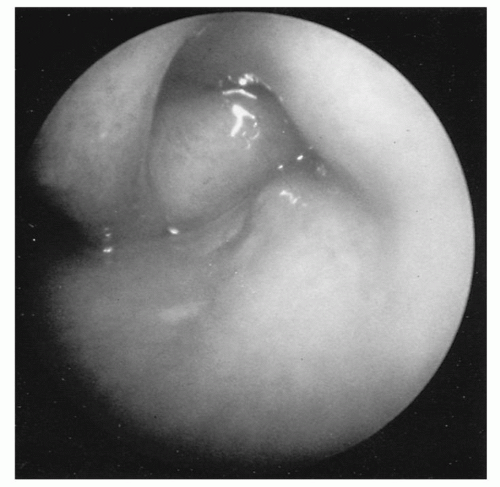 Figure 88.2 A nasolacrimal duct cyst may be identified under the inferior turbinate and may cause nasal obstruction. Such children often present with concomitant dacryocystitis. |
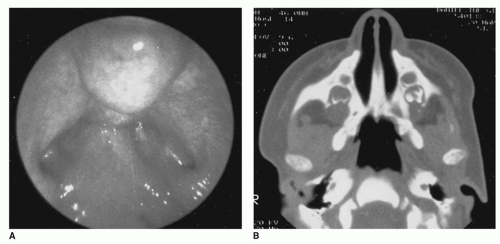 Figure 88.3 Choanal atresia may be identified endoscopically as seen in this view with a 120-degree telescope (A) and confirmed by CT scan (B). |
Regardless of the cause of obstruction, establishing a secure airway is paramount. In many circumstances, this must be done prior to any investigative procedures. If the child has no underlying lung disease or laryngotracheal abnormalities, placement of an oral airway may be sufficient. An alternative to the conventional oral airway may be a feeding nipple cut at the end. Either of these should be secured by taping or with tracheotomy ties placed around the head. Oxygen saturation monitoring can determine the efficacy of this maneuver and, if the child does not maintain adequate oxygen saturation levels, supplemental oxygen may be required. Alternatively, orotracheal intubation with possible ventilatory support may be required. In any child with airway obstruction, failure to maintain adequate ventilation with an endotracheal tube in place may signify intrinsic pulmonary disease, and the active assistance of a neonatologist should be obtained (1).
ORAL CAVITY/OROPHARYNX/HYPOPHARYNX
Examination of the mouth, oropharynx, and hypopharynx follows the nasal exam. Patients with obstruction in this area usually present with coarse inspiratory stertor that increases during sleep. The cry is usually normal, but may be muffled. Suprasternal and sternal retractions are common and may increase to total chest retractions with severe obstruction. Feeding may be difficult or impossible because of an anatomic obstruction. The airway noise is frequently worse with supine positioning, feeding, and agitation. If immediate intervention is needed, placement of an oral airway, nasopharyngeal airway, or endotracheal tube should be considered.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


