Chapter 17 Molecular Pathogenesis of Thyroid Neoplasia
Thyroid Neoplasia: An Overview
Epidemiology
Thyroid carcinoma accounts for about 1% of all malignancies and more than 95% of malignancies of the endocrine system.1 The age-adjusted incidence of thyroid cancer is 0.5 to 10/100,000 persons per year. In women, the incidence is about threefold that of men and the peak of incidence is one to two decades earlier than it is for men.1 In the United States, 44,000 new thyroid cancer cases and 1690 deaths were expected in 2010.2 The incidence of thyroid cancer, mainly small papillary carcinomas, has been increasing steadily since the 1980s.1,2 It is still unclear whether this trend reflects increased medical surveillance and more accurate diagnostic scrutiny or instead reflects a real increase because of the enhanced exposure to environmental risk factors, such as ionizing radiation or chemical carcinogens.3
Morphology
The thyroid gland is composed of two distinct hormone-producing cells: follicular cells (producing thyroid hormones) and parafollicular C cells (producing calcitonin). The majority of thyroid neoplasms originate from these two cell types.4 There are five major types of thyroid carcinoma: papillary (PTC), follicular (FTC), poorly differentiated (PDTC), anaplastic (ATC), and medullary (MTC) carcinoma (see Chapter 44, Surgical Pathology of the Thyroid Gland).4 PTC, FTC, PDTC, and ATC arise from follicular cells and are collectively designated NMTC (nonmedullary thyroid carcinomas). Medullary (MTC) thyroid carcinoma arises from the calcitonin-secreting parafollicular C cells.4,5 It is likely that thyroid tumors also arise from thyroid stem cells as has been demonstrated for many other tumor cell types.6 It was recently demonstrated that FTC, PTC, and ATC contain a small subpopulation of tumorigenic stemlike cells with high aldehyde dehydrogenase (ALDH) activity. These cells have unlimited replication potential and recapitulate the behavior of the parental tumor.7 Cells with stem cell-like properties have also been isolated from MTC.8
Papillary (PTC) and follicular (FTC) thyroid carcinomas maintain differentiated thyroid functions and are collectively referred to as differentiated thyroid carcinomas (DTC) (see Chapters 18, Papillary Thyroid Cancer, 19, Papillary Thyroid Microcarcinoma, 20, Follicular Thyroid Cancer, and 21, Dynamic Risk Group Analysis for Differentiated Thyroid Cancer). Poorly differentiated (PDTC) and anaplastic (ATC) are less differentiated or undifferentiated cancer types, respectively.4
One additional subtype of follicular cell–derived thyroid tumor that is worthy of special mention are the Hurthle cell tumors (see Chapter 22, Hurthle Cell Tumors of the Thyroid). Although initially Hurthle cell tumors were classified in a separate category, they are currently considered variants of differentiated thyroid tumor types that are distinguished by an accumulation of Hurthle (also called Askanazy) oncocytes.9 Oncocytes are large polygonal cells with voluminous granular eosinophilic cytoplasm because of the accumulation of mitochondria.9 This accumulation is believed to be compensatory and secondary to defective oxidative phosphorylation and ATP synthesis. Mutations in mitochondrial genes coding for complexes I and III of the respiratory chain, as well as of the nonmitochondrial GRIM-19 gene that controls mitochondrial membrane potential, have been detected in oncocytic thyroid tumors.9 It is worth mentioning that oncocytic changes are also commonly seen in non-neoplastic conditions like thyroiditis, multinodular goiter, or Graves’ disease.
Prognosis
ATC is invariably fatal.1,10 DTC often has an indolent clinical course with low morbidity and mortality.1 However, some patients with DTC may have a recurrence or develop de-differentiated radiodine-refractory tumors, and these tumors generally have a worse prognosis (see Chapters 18, Papillary Thyroid Cancer, 19, Papillary Thyroid Microcarcinoma, 20, Follicular Thyroid Cancer, 21, Dynamic Risk Group Analysis for Differentiated Thyroid Cancer, and 26, Anaplastic Thyroid Cancer and Thyroid Lymphoma). Although primary surgery is curative in the majority of MTC patients treated at an early stage, disease can persist or recur, and local and distant metastases are the major causes of mortality for MTC.5 For radioiodine-refractory DTC, ATC, and metastatic MTC, novel therapeutic measures are urgently needed.
Inheritance
Approximately 25% of MTC cases are inherited.11 NMTC, mainly PTC, can also be of familial origin (5% of PTC cases) (see Chapter 29, Familial Nonmedullary Thyroid Cancer). Familial risk associated with NMTC is among the highest of all solid tumors.12,13 Familial NMTC (FNMTC) can be subdivided into two groups: syndromic and nonsyndromic FNMTC.12,13 Although syndromic FNMTC patients may develop different tumor types, nonsyndromic FNMTC patients develop primarily thyroid tumors. Syndromic FNMTC is associated with rare Mendelian tumor syndromes, like Cowden’s disease, Carney’s complex, paraganglioma syndrome, familial adenomatous polyposis (FAP), and Werner syndrome (progeria).13 FNMTC features a polygenic mode of inheritance, and some genetic loci have been proposed as important for FNMTC.12 Variants of two genes, FOXE1 (TTF2) on 9q and NKX2-1 (TTF1) on 14q, both coding for thyroid-specific transcription factors, confer an increased risk of thyroid cancer.14 It is anticipated that these findings will soon be translated into better screening for those at risk of developing FNMTC.
Genetics
The different morphologic subtypes of thyroid cancer correlate with specific genetic lesions (Table 17-1).15,16 This knowledge has provided the theoretic basis for clinical studies aimed at exploring the detection of such genetic lesions to improve diagnosis17 and to refine prognosis,18 as well as for clinical trials aimed at testing molecularly targeted agents to improve therapy of thyroid cancer patients.19 A recent study demonstrated that detection of thyroid cancer–associated genetic lesions may help refine the diagnosis of thyroid nodules based on FNA. In particular, the detection of mutations in the BRAF, RAS, RET/PTC, and PAX8-PPARG genes was a strong indicator of cancer: 97% of mutation-positive nodules had a malignant diagnosis on final histopathologic review.17
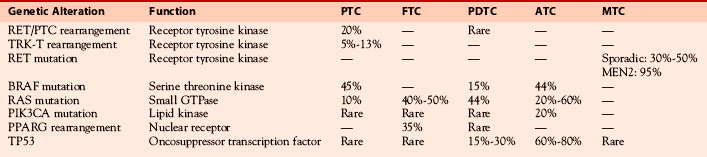
Because many of the known mutations contributing to thyroid cancer affect protein kinases, which are amenable to interference with small molecular inhibitors that are able to compete with ATP,20 this strategy is currently being pursued in phase II and III clinical studies (see Chapter 55, Medical Treatment for Metastatic Thyroid Cancer). Initial reports from studies treating thyroid cancer patients with ATP-competitive inhibitors that block RET, BRAF, or other kinases suggest encouraging early results.19
In addition to mutations in protein-encoding genes, recent reports have strongly suggested that microRNA genes also have an important role in the molecular pathogenesis of thyroid cancer.21 MicroRNAs (miRNAs) are short (20 to 24 nt) noncoding RNAs that are involved in the posttranscriptional regulation of gene expression by affecting either stability or translation of messenger RNAs. miRNAs are synthesized as primary transcripts (pri-miRNAs) that in turn are cleaved by the Drosha ribonuclease to produce 70-nt stem-loop precursor miRNAs (pre-miRNAs). Pre-miRNA is further cleaved by the Dicer ribonuclease to generate the mature miRNA.21 The mature miRNA is incorporated into an RNA-induced silencing complex (RISC), which recognizes target mRNAs. Specific sets of miRNAs appear to be either up- or down-regulated depending on the type of tumor, and this may lead to the identification of novel diagnostic markers and therapeutic targets.21
Thyroid Neoplasia: Genetic Alterations Associated with Specific Thyroid Tumors
Follicular Adenoma
Thyroid nodules are very common and can be found at palpation in 5% to 20% of unselected subjects. With modern ultrasonographic techniques, up to 67% of persons may harbor thyroid nodules ≥ 2 mm.22 These nodules are rarely malignant (less than 5%), and benign nodules can be colloid nodules, cysts, inflammatory nodules, hyperplastic nodules, or adenomas (follicular thyroid adenoma [FTA]) (see Chapter 11, The Evaluation and Management of Thyroid Nodules). FTAs can be either hypo- (cold) or normo-functioning (warm) adenomas or hyperfunctioning (hot or toxic) adenomas. Hot nodules are associated with distinct genetic alterations compared to cold or warm nodules.15
Thyroid-stimulating hormone (TSH) binds to its seven-transmembrane-domain G-protein-coupled receptor (TSHR) and activates the GSα–adenylyl cyclase–cyclic AMP (cAMP) cascade; in turn, cAMP stimulates follicular cells to grow and produce thyroid hormones (Figure 17-1).23 Hyper-functioning thyroid adenomas are typically caused by gain-of-function mutations that hijack the TSH-cAMP signaling pathway. These lesions comprise mutations of TSHR (80% of cases) and of GNAS1 (which encodes GSα) (6% of cases).23 TSHR mutations are also found in familial nonautoimmune hyperthyroidism. In contrast, mutations of TSHR and of GNAS1 are virtually absent in malignant tumors, suggesting that overactivation of this pathway does not lead to malignant transformation.15
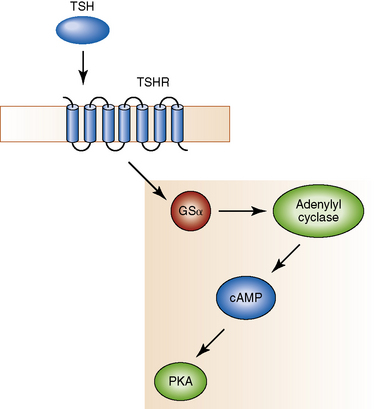
FTAs that are warm or cold do not bear mutations in the TSHR-cAMP cascade. These FTAs (like hot nodules) share cytologic features with FTC. FTA and FTC are distinguished chiefly by the presence of invasion beyond the tumor capsule or into blood vessels.4,22 The distinction between FTA and FTC can sometimes be difficult, even on final histopathologic review, and some follicular lesions are characterized as minimally invasive follicular carcinomas (see Chapter 44, Surgical Pathology of the Thyroid Gland). The apparent morphologic continuum between follicular adenomas and invasive follicular carcinomas suggests that for some follicular lesions there is a progression from benign to malignant tumors. Molecular findings support this adenoma to carcinoma sequence in follicular thyroid lesions. FTAs feature the same genetic alterations that are found in FTC, though with a lower prevalence. RAS mutations are found in about 40% to 50% of FTC and 20% to 40% FTA (see Table 17-1). The presence of RAS mutations has been correlated with a less favorable prognosis in FTC.16 Thus, although not specific for malignancy, RAS mutation detection in FNA samples may suggest that that lesion is at higher risk of malignant progression.16 Likewise, the prevalence of PPARG rearrangements is lower in FTA (2% to 10%) than FTC (35%).16,24 Molecular determinants of malignant conversion of FTA into FTC remain unknown.
Papillary Thyroid Carcinoma
PTC is the most prevalent thyroid cancer subtype (80% of cases).1,4 PTC is characterized by typical features of the cell nuclei, including enlarged, vesicular, and overlapping nuclei with clearing, fine dusty chromatin, grooves, inclusions, and single or multiple micro- or macronucleoli. PTC is closely linked to radiation exposure, and its incidence sharply increased in children after the Chernobyl accident (see Chapter 28, Chernobyl and Radiation-Induced Thyroid Cancer).25
PTC can be subdivided into several subtypes, the most common types being classic variant PTC (CV-PTC), follicular variant PTC (FV-PTC), and tall cell variant PTC (TCV-PTC). TCV is an aggressive PTC variant, characterized by at least 50% large granular eosinophilic tumor cells, at least twice as long (tall) as they are wide, and with multiple intranuclear inclusions.26
Over 70% of PTC feature genetic alterations causing activation of the MAPK pathway.15,16 This pathway is typically initiated by the activation of a membrane-bound growth factor receptor (receptor tyrosine kinase [RTK]) (Figure 17-2). Thanks to their intrinsic kinase activity, activated growth factor receptors autophosphorylate various cytoplasmic tyrosines, which then become binding sites for intracellular molecules containing phosphotyrosine-binding motifs.27 Recruitment of Grb2-SOS complexes to the activated RTK leads to GTP exchange on RAS, a small GTPase. Binding to RAS in its GTP-bound state promotes the activation of the RAF family of serine/threonine kinases, which includes c-RAF, BRAF, and ARAF. Once activated, RAF kinases phosphorylate MEK (MAP kinase or ERK kinase), which, in turn, phosphorylates and activates MAPK (or extracellular signal-regulated kinases [ERKs]).27 As Table 17-1 shows, PTC is characterized by mutations of various components of the MAPK, including rearrangements of the RET tyrosine kinases, or more rarely NTRK1, or activation of BRAF. RET/PTC and BRAF alterations are mutually exclusive in PTC, suggesting that mutations at more than one of these sites are unlikely to confer additional biologic advantage, a fact that is consistent with the overlap of the signaling pathways of these oncoproteins (see Figure 17-2).
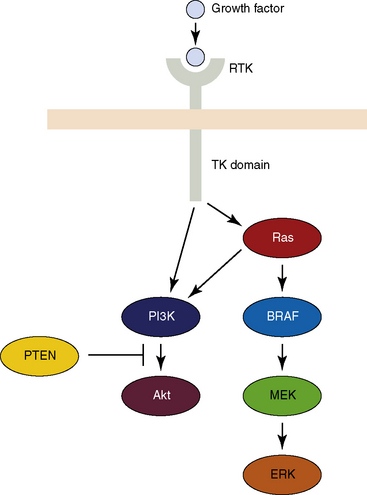
The genetic lesions that are typical of PTC are thoroughly described later. It can be anticipated that specific PTC subtypes have specific mutation patterns. For example, FV-PTC, when encapsulated, frequently (up to 40%) may harbor mutations in RAS.28 In contradistinction, infiltrative FV-PTC often (25%) harbor BRAF mutations. BRAF mutant FV-PTC may harbor the K601E or other alternative mutations rather than the classical BRAF V600E. Compared to FV-PTC and classical PTC, TCV-PTC show a high (up to 80%) proportion of BRAF V600E positive cases.16
PTC has been associated with the deregulation of a specific set of microRNAs.21 Up-regulation of miR-221, -222, -146b, and 181b was consistently reported in different PTC series.29 Intriguingly, miR-221 and -222 target, among the others, the cell cycle inhibitor p27kip1. Thus, their up-regulation in PTC favors the down-regulation of p27kip1 and therefore the loss of cell cycle control.29
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


