Molecular Genetic Basis of Eye Diseases
Tracy W. Patton
Ming X. Wang
In the last 10 years, we have seen an explosion of knowledge in the understanding of molecular mechanisms of disease processes as a result of cloning of a number of important disease-causing genes, such as those for cystic fibrosis,1 familial breast and ovarian cancer,2,3 colon cancer,4 Huntington disease,5 Alzheimer disease, and obesity.6 Because vision is such an important sense, and because many genetic abnormalities that affect visual function can be detected, research on the molecular basis of ophthalmic diseases has grown tremendously in recent years. In fact, many important discoveries in molecular ophthalmology have played critical roles in the development of entirely new fields in molecular genetics. For example, the identification and cloning of the retinoblastoma gene (RB1) has served as the prototype of an important new class of cancer-causing genes: the tumor-suppressor genes. In retinitis pigmentosa, DNA point mutations have been identified, serving as the first example of retinal degeneration caused by DNA alterations in photoreceptor proteins. In many ways, the study of the molecular basis of ocular diseases has led to many exciting new discoveries in the molecular biology of human diseases in general.
Faced with these rapid new developments in molecular ophthalmology, it is becoming imperative that ophthalmologists who are involved in the day-to-day care of patients keep abreast of the major scientific advances in the field and that they recognize new diagnostic and therapeutic modalities. As of 1996, many hereditary ophthalmic diseases have been examined in detail through the use of molecular genetic techniques. In many instances, the actual disease genes have been identified and mutations characterized in patients. This chapter provides a review of the basic principles of molecular genetics as applied to ophthalmology as well as a discussion of the current understanding of molecular pathogenic mechanisms of many important ocular diseases. Clinical applications are described, including DNA diagnosis, genetic counseling, and treatment. In an attempt to help ophthalmologists grasp the essential features of these exciting new scientific developments, this chapter places emphasis on the correlation between laboratory findings and clinical disease entities. Excellent reviews of this field can also be found elsewhere.7,8,9,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25
Principles of Molecular Ocular Genetics
Patterns of Inheritance of Ophthalmic Diseases
Out of the 4,000 genetic diseases that affect humans, nearly 33% have ocular manifestations.26 These diseases can be transmitted according to various modes of genetic transmission, including autosomal dominant, autosomal recessive, X-linked dominant (rare) or recessive, multifactorial inheritance, and cytoplasmic inheritance.
The most common mode of transmission for ocular diseases is autosomal-dominant transmission. Examples of ocular diseases with autosomal dominant traits include aniridia, Best disease, corneal dystrophies, retinoblastoma, and neurofibromatosis. For an inherited disorder to be classified as autosomal dominant, it must meet four general criteria27:
Affected members appear in every generation, each patient having an affected parent.
Any child of an affected parent has a 50% risk of inheriting the trait.
Phenotypically normal family members do not transmit the phenotype to their children.
Males and females are equally likely to transmit the phenotype to children of either sex.
Exceptions to these rules, however, do exist. For example, an autosomal-dominant disorder may have a reduced penetrance, which is defined as the percentage of persons with a given genotype who are affected. Studies of low-penetrant disorders have provided insight into the molecular pathogenicity of various DNA mutations, as demonstrated in the case of retinoblastoma.28,29,30,31
The criteria for a disorder to be classified as autosomal recessive are as follows27:
It appears in more than one member of a kindred and typically is seen only in the sibship of the proband, not in parents or offspring.
The recurrence risk for each sibling of the proband is 25%.
The parents of the affected person often are consanguineous.
Males and females are equally likely to be affected. Autosomal-recessive disorders in ophthalmology include gyrate atrophy, Stargardt disease, Tay-Sachs disease, and Usher syndrome.
X-linked recessive disorders are the most common of the X-linked disorders. Examples of X-linked recessive disorders in the eye include choroideremia, ocular albinism, red-green color blindness, and Fabry disease. X-linked recessive disorders are characterized by the following five criteria27:
The incidence of the trait is much higher in males than in females.
The disease gene is transmitted from an affected man through all of his daughters. Any of his daughters’ sons have a 50% chance of inheriting the disease gene.
The gene is never transmitted directly from father to son.
The gene may be transmitted through carrier females.
Heterozygous females are usually unaffected, but some may express the condition with variable severity.
Other modes of genetic transmission include multifactorial inheritance and cytoplasmic inheritance. Multifactorial inheritance is characterized by the presence of a significant environmental influence. Many ophthalmic diseases such as age-related macular degeneration and primary open-angle glaucoma may eventually prove to be in this category. Examples of cytoplasmic inheritance include mitochondrial disorders, such as Leber hereditary optic neuropathy and Kearn-Sayre syndrome. These traits are characterized by a maternal transmission pattern.
Gene Structure and Function
The physical basis of a gene is the DNA molecule. Since the demonstration of the DNA structure in 1953 by James Watson and Francis Crick, the DNA molecule has become the cornerstone of molecular biology. A DNA molecule consists of four basic building blocks: adenine (A); guanine (G); cytosine (C); and thymine (T). The remarkable precision by which A is paired only with T, and G only with C ensures genetic reproducibility. In the nucleus of a cell, the DNA molecule is transcribed into a RNA molecule, which differs from DNA in that RNA is single-stranded and contains uracil (U) instead of thymine (T). The messenger RNA (mRNA) molecule is then translated into a protein molecule on ribosomes in the cytoplasm, completing the process of gene expression. Alterations at the level of DNA can be minute (as in point mutation of a base) or more extensive (as in a deletion of a large segment of DNA). These DNA alterations can result in the production of abnormal RNA molecules, leading to protein abnormalities and human diseases.
The Human Gene Map
In recent years, the Human Genome Project has made remarkable progress, mainly due to the development of new cloning and sequencing strategies, such as yeast artificial chromosomes and microsatellite repeat markers.32,33 A map of 30,181 human gene-based markers has been integrated within the current genetic map by radiation hybrid mapping. This new map contains twice as many genes as the previous map and is two to threefold more accurate.34 The availability of a human gene map will provide molecular biologists with a road map that not only will greatly facilitate the search for disease-causing genes, but will also make possible rational design of genetic diagnosis and therapy.
Experimental Techniques Used in Molecular Genetics
In the following sections, essential features of major experimental tools used in molecular biology will be described, particularly with regard to studies of genes related to vision.
Polymerase Chain Reaction
Invented in 1988,35 the polymerase chain reaction (PCR) has revolutionized molecular biology, particularly with regard to gene amplification and cloning. The goal of PCR is to amplify a DNA fragment of interest in sufficient quantity for molecular biologic analysis. In a typical PCR reaction, the parent DNA fragment is bound by two primers—short stretches of DNA molecules with sequences complementary to either terminus of the parent DNA fragment. A thermally stable DNA polymerase is then added, and the reaction mixture undergoes a series of repeated thermal cycles. The number of copies of the product DNA fragments will then be amplified to 2n, where n is the number of amplification cycles. PCR can typically amplify DNA fragments up to 1 kilobase (kb) in size. To amplify larger DNA fragments, one has to resort to gene cloning using vectors.
Gene Cloning Using Vectors
Cloning of large gene fragments requires the use of a vector. A vector is typically a circular, double-stranded DNA molecule such as a bacterial plasmid. Using restriction endonuclease enzymes, a target DNA fragment of interest is inserted into the circular DNA structure of a plasmid vector, thus creating a recombinant DNA clone. The clone is then transformed into a host bacterial cell and is amplified along with the cell divisional process of the host. Finally, multiple copies of the original recombinant clone are then isolated from the host bacteria cells. The goal of the entire cloning process is to amplify the original gene fragment for further molecular characterization. There are also new ligation-independent cloning methods that allow for rapid and seamless assembly of vectors from component modules. Now, gene cloning can be accomplished simultaneously with the assembly of a modular vector. This will allow for more flexibility in the cloning of genes into custom assembled modular vectors.36
Southern Blot
Invented by Edwin Southern in 1975,37 the Southern blot is used to separate DNA fragments by electrophoresis and then to transfer the fragments onto a solid support. A DNA sample to be analyzed is first digested with a restriction endonuclease, which cuts the DNA at certain specific sequences. These fragments are separated by size with the use of gel electrophoresis. The gel electrophoretic pattern is then transferred onto a solid support, such as nitrocellulose. A DNA probe is then constructed with its sequence complementary to that of the target DNA fragment to be visualized. The DNA probe is radioactively labeled and hybridized with the target DNA fragments immobilized on the nitrocellulose filter. The final electrophoretic pattern of only the DNA fragment of interest (as picked out by the specific DNA probe) is visualized by autoradiography. The power of the Southern blot lies in its ability to characterize the sizes of DNA fragments containing specific sequences. The technique can be used, for example, to detect large deletions or duplications of a gene fragment, as is sometimes found in patients with retinoblastoma.
Restriction Fragment Length Polymorphism
In 1980, Botstein and associates38 discovered that the naturally existing degree of variation in the distribution of restriction enzyme sites found in human chromosomes can be used as landmarks for identifying the chromosomal location of gene segments. This finding, termed restriction fragment length polymorphism (RFLP), has greatly accelerated linkage analysis and the characterization of human genes. In an RFLP analysis, DNA from the patients is cleaved by restriction enzymes, and the resulting DNA fragments are separated by gel electrophoresis. Because each patient has a specific distribution of these restriction enzyme sites, the RFLP pattern is unique to each patient. Currently several hundred restriction enzymes are available for RFLP analysis. The combination of a collection of RFLP markers can often be much more effective in detecting the presence or absence of a specific DNA fragment in a particular person, as demonstrated in the genetic diagnosis of retinoblastoma.30,39
Variable Number of Tandem Repeats
For a DNA marker to be useful in characterizing unique DNA fragments, it should have a large number of polymorphic forms. An RFLP marker often has only two polymorphisms, thus limiting its use in genetic testing. Another class of more powerful DNA markers has since been found: the variable number of tandem repeats (VNTR) marker.40 A VNTR marker consists of a series of allelic fragments that are related to each other by a variable number of repeats of a short stretch of DNA sequence (tandem). VNTR markers are highly polymorphic, making them the most commonly used DNA markers in the characterization of human chromosomal fragments. Several thousand VNTR markers have been identified to date throughout the human genome.41,42
Single-Strand DNA Gel Electrophoresis
This technique is used to detect the presence of DNA mutations in a given gene. A single-stranded gene fragment moves with a specific motility corresponding to its three-dimensional conformation determined by its sequence. If a gene fragment contains mutations, it will have an altered three-dimensional structure and thus a different electrophoretic motility compared with that of a normal gene. This method, termed single-strand conformational polymorphism (SSCP), is used for rapid screening of gene fragments to determine whether mutations are present.43 Figure 55.1 shows an example of the SSCP technique. Although the technique is efficient for detecting the presence of a mutation, it usually cannot determine the nature of the mutation. The exact alterations in base sequences of the gene fragments can be determined by techniques such as DNA sequencing.
 Figure 55.1. A diagram demonstrating two molecular detection techniques. Allele-specific hybridization (top) for the detection of point mutations in the rhodopsin gene in patients with autosomal-dominant retinitis pigmentosa. The dark spots represent DNA containing mutations at codon 23 of the rhodopsin gene. Electrophoretic pattern (bottom) demonstrating the nonradioactive single-strand conformational polymorphism technique. The leftmost lane corresponds to a retinoblastoma gene fragment containing a point mutation, resulting in altered motility compared with the rest of lanes with normal DNA. (Adapted from Wang MX, Donoso LA: Gene research and the eye. Curr Opin Ophthalmol 4(III): 102, 1993) |
DNA Sequencing
The discovery in 1977 of rapid DNA sequencing technologies by Sanger and colleagues44 and Maxam and Gilbert45 has helped usher in a new era of molecular biology. It became possible to determine the exact composition of genes with the ultimate resolution: the base-to-base DNA sequence. The Sanger method44 uses an ingenious chain-termination technique such that the target DNA fragment to be analyzed is replicated in small fragments that terminate at specific bases. The DNA sequence is then determined by the electrophoretic pattern of these DNA fragments. In contrast, the method used by Maxam and Gilbert45 cleaves the target DNA fragments into small pieces using chemical reagents that cut only at specific bases. The final DNA sequence is read from electrophoretic gel patterns of the cleaved DNA fragments.
Northern Blot
The Northern blot is the counterpart of the Southern blot. It is used to analyze RNA samples. The purpose of the northern blot is to determine the size and abundance of mRNA of a specific gene. RNA from a particular cell type is electrophoretically separated on a gel and transferred onto a solid support. To visualize these mRNA fragments, a DNA probe containing part of the sequence of the gene is constructed. The probe is radioactively labeled and hybridized with the RNA sample immobilized on the solid support. The resulting radioactively labeled target mRNA fragment is then analyzed by autoradiography.
Western Blot
A Western blot is used to analyze proteins. It is a counterpart of the Southern blot (for DNA analysis) and northern blot (for RNA analysis). A protein extract obtained from cells of a patient with a genetic disease is separated by size with the use of polyacrylamide gel electrophoresis. After transferring the protein fragments onto a solid support, radioactively labeled antibodies that specifically bind to the protein of interest are incubated with the protein fragments. The western blot is used to obtain information about the size and abundance of mutant proteins in patients suffering from a genetic disease.
DNA Mutations and Ophthalmic Diseases
Gene mutations lead to human diseases. A DNA mutation is defined as a permanent change in the sequence of a gene. There are three general categories of DNA mutations:27 genome mutations, chromosome mutations, and gene mutations. Genome mutations involve missegregation of a chromosome during cell division, as seen in Down syndrome (trisomy 21). Genome mutations are the most common type of DNA mutations seen in humans. Chromosome mutation occurs less frequently and involves rearrangement of chromosomes. An example of a chromosomal mutation in ophthalmology is the sporadic form of aniridia involving chromosome 11p deletion. In addition to aniridia, these patients may have a predisposition to Wilms tumor, genitourinary anomalies, and mental retardation.46,47,48 Gene mutation is caused by errors in DNA replication or induced mutation by mutagens.
Human DNA replication is highly efficient and accurate. Spontaneous errors in DNA replication occur only about once every 10 million base pairs.27 A wide variety of chemical and environmental mutagens, however, can cause induced gene mutations. Gene mutations can involve deletions and insertions of gene segments, or point mutations that involve a single nucleotide substitution. A point mutation can alter a triple base codon, resulting in a change in an amino acid in a protein product. A single nucleotide substitution can be either a transition mutation, in which one purine is changed to another purine (e.g., A changed to G), or a transversion mutation, in which a purine is replaced by a pyrimidine or vice versa (e.g., A replaced by C, or C by A).
Gene mutation can occur in a variety of forms, including missense mutation, nonsense mutation, or RNA-splicing mutation. A missense mutation is a point mutation in a DNA sequence that alters a triplet code for an amino acid, resulting in the replacement of an amino acid. In autosomal-dominant retinitis pigmentosa (ADRP), for example, the first point mutation identified in the rhodopsin gene consists of the replacement of C by A, which results in the change of the amino acid proline to histidine.49 Because proline is important in the three-dimensional structure of rhodopsin, its loss leads to a functional alteration of the protein. Nonsense mutation consists of a DNA point mutation that creates a stop codon in the corresponding mRNA, resulting in the premature termination of the translation of the protein and giving rise to a shortened or truncated protein. Examples in ocular diseases include a nonsense mutation in the rhodopsin gene in patients with autosomal-recessive retinitis pigmentosa50 and premature termination codon identified in patients with Stickler syndrome (arthro-ophthalmopathy; see Stickler Syndrome section later in chapter). In RNA splicing mutation, transcription of DNA produces unprocessed RNA, which is subsequently spliced to remove the intron sequences, leaving only exons. DNA point mutations involving bases corresponding to the RNA-splicing region may result in abnormal processing of RNA. An example of an RNA-splicing mutation involving ocular disease can be seen in retinoblastoma. As demonstrated by Horowitz and co-workers,51 a point mutation at the splicing site of exon 21 of RB1 causes the loss of exon 21 in the mRNA product, resulting in a truncated retinoblastoma protein.
The relationship between the types and locations of DNA mutations and the extent of clinical manifestation of human diseases is an active area of research today. Advances in this field not only will improve our understanding of the molecular mechanism of the disease processes, but also will greatly aid in DNA diagnosis and genetic counseling. More than 40 types of RB1 mutations have been identified. Notably, more than one third of these mutations occur in the noncoding region of RB1, grossly altering the RB1 product by affecting processes such as RNA splicing. Among the DNA mutations identified in RB1, there appears to be no “hot spot” (i.e., a point mutation that occurs in a majority of the patients). This is in contrast to other human diseases (e.g., cystic fibrosis), in which a single point mutation accounts for 70% of patients with the disease (a three base pair deletion that removes the phenylalanine at position 508 of the cystic fibrosis gene [CF]).1,52,53,54 This is particularly relevant in clinical DNA diagnosis and genetic counseling, since the lack of a hot-spot mutation in RB1 makes it necessary to screen and analyze all segments of RB1 in order to identify the causative mutation, a time-consuming and laborious process.
Genetic Heterogeneity
The clinical manifestations of genetic mutations depend on many factors. The following are several key concepts in examining the relationship between genotypes (DNA mutations) and phenotypes (clinical disease).24
Allelic heterogeneity refers to the concept that various mutations in one gene can be associated with different clinical presentations of the disease. For example, in the human rhodopsin gene, different mutations are found, which are associated with varying clinical severities of ADRP.
Nonallelic heterogeneity (locus heterogeneity) refers to the observation that mutations in several different genes can be associated with various clinical phenotypes of the disease. For example, several gene mutations associated with ADRP have been identified, including mutations of the rhodopsin and human peripherin/RDS genes (located on chromosomes 3q and 6p, respectively).
The concept of expressivity refers to the extent of disease manifested in patients who have the same gene mutation. For example, in some cases of ADRP, family members sharing the same inherited rhodopsin gene mutation show different degrees of the disease.55 Another example is in the case of familial exudative vitreoretinopathy (FEVR). FEVR can be inherited as an autosomal-dominant disease with almost full penetrance. The severity of the disease, however, varies significantly, even among family members in the same pedigrees who have inherited the same gene mutation. Some patients may be found to have minimal disease, discovered only after a more severely affected member of the family is examined.56
Penetrance refers to the percentage of family members in a given pedigree who have inherited the same mutant allele and manifest the disease clinically. In retinoblastoma, it has been noted that certain mutant retinoblastoma alleles have a lower penetrance than that expected from a classic autosomal-dominant transmission.28,29,30,31
Identification of Genes Involved in Ocular Diseases
There are two main strategies in mapping and identifying disease-causing genes: positional cloning and functional cloning.
Positional Cloning and Linkage Analysis
In positional cloning and linkage analysis, DNA segments are examined to see whether a set of DNA markers cosegregates with affected persons. The rapid development in the Human Genome Project and the availability of the second- and third-generation human genome markers, with a resolution of 2 centimorgans or less, has rapidly advanced the speed and accuracy of linkage analysis.
In a typical chromosomal linkage study, statistical analysis is performed to determine the probability of cosegregation of the DNA markers with the disease. The closer the chromosomal distance between a DNA marker and a disease gene, the more likely it is that the DNA marker will travel together (cosegregate) with the disease gene in chromosomal recombination events. A score of the logarithm of the odds favoring linkage (LOD score) is used to quantify the distance between the marker and the disease gene. Because the LOD is calculated as the logarithm to base 10 of the odds in favor of linkage, a LOD score of 3 represents a probability of 1000:1 that the observation of linkage did not occur by chance and is often taken as the minimal LOD value for presumptive evidence of linkage. In addition to helping identify the disease gene, these DNA markers (particularly intragenic DNA markers) have also proved useful for clinical genetic diagnosis.11,18,20,30,39,57,58,59,60,61,62,63 The above-described strategy of linkage analysis and positional cloning is often termed “reverse genetics.” This is because, in contrast to certain examples in classical genetics such as sickle cell anemia, in which the biochemical defect is known before the chromosomal localization of the genetic defect, in linkage analysis and positional cloning there is no prior knowledge of the nature of the biochemical defect involved in the disease.
Positional cloning typically consists of two general strategies, chromosome walking and chromosome jumping. For example, in the cloning of CF, linkage studies mapped the disease gene locus to chromosome 7q31. Chromosome walking techniques were then used to reveal the finer details in 7q31 region. Chromosome walking involves first creating a library of clones by cleaving the entire genome and cloning these fragments. A DNA marker in the chromosomal region established by linkage analysis is then used to “fish out” a clone in the library that cosegregates with the disease. Once such a clone is identified, it is cleaved into smaller fragments, which are subcloned. One of the subcloned fragments is then regarded as the “first step” in chromosome walking. It is then used as a DNA probe to fish out the next adjacent clone in the library, walking onto the “next step” and thus organizing the clones in a series. This chromosome walking process is repeated until the disease gene is finally encountered. In the case of cystic fibrosis, exhaustive chromosome-walking effort has converged on a DNA segment that might contain CF. The gene tracking effort was then complemented and accelerated by a second technique, chromosome jumping. Chromosome jumping relies on the creation of larger DNA clones and recircularization processes to track down genes. With the combination of chromosome walking and jumping techniques, a DNA segment that appeared to contain CF was finally isolated.1,52,53,54
The identification of a disease-causing gene must satisfy the following requirements, as demonstrated in cystic fibrosis1:
The putative gene should contain an open reading frame with the proper start and stop regions expected of a gene.
It should contain CG-rich sequences in the 5′ upstream region, consistent with a regulatory sequence structure of a gene.
It should be evolutionarily conserved, as demonstrated by “zoo” blot (comparison of a DNA fragment among species).
It must have a transcript (i.e., the corresponding mRNA is present in a cell).
Once a disease gene is identified, verification of its authenticity is important. Again let us discuss cystic fibrosis as an example. The putative CF satisfies the following criteria:
Mutations in the putative CF have indeed been found in patients with the disease.
The protein structure encoded by CF resembles that of a membrane transport protein, consistent with the expectation based on the pathophysiologic abnormality.
Transfection of the intact CF corrects the defect in animal models.
Linkage analysis and positional cloning, although extremely powerful, have limitations. For instance, for a disease with locus heterogeneity such as that shown in many hereditary retinal diseases, linkage data may be susceptible to error, because entirely different gene loci may be deranged in a single disease. This is the case for ADRP, where at least 10 different genetic loci have been identified to date.14,20,24,49,64,65,66,67,68,69,70,71,72,73,74,75,76,77,78,79,80,81,82,83,84,85,86,87,88,89,90,91,92,93,94 In addition, linkage analysis and positional cloning are labor intensive in that they both require extensive analysis of DNA markers. In some instances an alternative strategy, functional cloning, may offer a short cut to uncover the genetic defect.
Functional Cloning (The Candidate Gene Approach)
The second major strategy for localizing disease-causing genes is called functional cloning (or candidate gene approach), in which a gene located in the chromosomal region of interest is chosen for study because it is likely to be related to the disease process. If mutations indeed are found in this candidate gene in patients who have a given disease, the candidate gene is then “elected.” With the rapid accumulation of DNA sequence data from the Human Genome Project, it has become increasingly feasible to make use of the candidate gene approach. Three main guidelines govern the candidate gene approach:
The location of the candidate genes should be consistent with prior linkage studies.
The biologic function of the candidate gene should be logically related to the disease process.
The candidate gene product should be expressed in the tissue examined.
The successful demonstration of the first point mutation in the rhodopsin gene in ADRP49 is an excellent example of the candidate gene approach. It was made possible by a prior study by McWilliam and colleagues,64 who demonstrated segregation of retinitis pigmentosa in a large Irish pedigree to chromosome 3q (i.e., the same chromosomal location as rhodopsin). Examination of the rhodopsin genes in patients with ADRP did reveal causative DNA mutations.49
Molecular Basis of Ophthalmic Diseases
In Table 55.1, we have provided a list of the chromosomal locations of several ocular diseases. In the following sections, we will discuss specific ocular diseases for which progress has been made recently regarding the molecular pathogenesis.
TABLE 55.1. Chromosomal Locations of Disease Genes With Ocular Manifestations | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
Albinism
Albinism constitutes a group of inherited disorders characterized by defective melanin synthesis and decreased skin, hair, and ocular pigmentation (Fig. 55.2).99 Ocular manifestations include iris transillumination, foveal hypoplasia, photophobia, nystagmus, and a severe decrease in visual acuity. There are two types of albinism: ocular and oculocutaneous.95,96,97 Ocular albinism-can be either X-linked recessive or autosomal recessive; oculocutaneous albinism can be autosomal recessive or genetically heterogeneous. Recently, significant advances have been made in understanding the molecular biology of albinism, which has allowed genetic diagnoses on the basis of carrier detection and prenatal genetic testing.
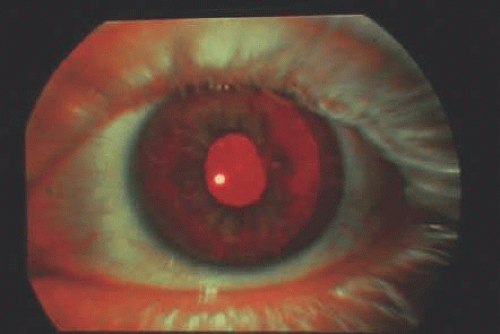 Figure 55.2. Ocular albinism. Decreased skin and hair pigmentation and iris transillumination defect can be seen. |
X-linked ocular albinism is characterized by the presence of macromelanosomes and it is divided into two types: type 1 (Nettleship-Falls type) and type 2 (Forsuus-Erikssontype).98 The mutations are in the genes involved in the biosynthesis of melanin pigment.99 These patients demonstrate foveal hypoplasia, hypopigmentation of the retina and decreased visual acuity. The skin pigmentation, however, is normal. The OA1 gene has been mapped to chromosome Xp22.2- p22.3,100 whereas the OA2 gene is located in the Xp21.3-21.2 region.101
There are two types of oculocutaneous albinism: tyrosine-negative (type 1) and tyrosine-positive (type 2). Patients with tyrosine-negative (type 1) oculocutaneous albinism typically have no pigment in their hair at birth, and the amount of pigment in the skin and eyes is scarce. The OCA1 gene locus has been mapped to chromosome 11q14-21, where the tyrosinase gene is located. The human tyrosinase gene consists of five exons. The length of genomic DNA is >50 kb.102 Tyrosinase is initially translated into a 529-amino-acid polypeptide, which is subsequently cleaved to produce the mature tyrosinase protein.103,104 Various mutations have been detected in the tyrosinase gene in patients with oculocutaneous albinism type 1.105,106 The most common type of mutation is a single base substitution that gives rise to a different amino acid in the tyrosinase protein product.97 A striking feature of patients with these tyrosinase mutations is that most of them are compound heterozygotes with different mutant alleles, hence the wide range of tyrosinase expression and the corresponding severity of oculocutaneous albinism type 1 seen clinically.
Clinically, patients with tyrosine-positive oculocutaneous albinism -(OCA2) have pigmented hair at birth, and the amount of pigmentation in the skin and eyes increases with age. The OCA2 gene locus has been mapped to human chromosome 15q11.2-q12. The OCA2 gene has been shown to be a homologue of the mouse pink-eyed dilution gene. The OCA2 gene product appears to function as part of melanosomal membrane and is involved in the transport of tyrosinase.
Clinical diagnosis of oculocutaneous albinism using these findings is proving to be feasible. One of the major difficulties in the molecular genetic screening and prenatal diagnosis of oculocutaneous albinism type 1 is that there appears to be no hot-spot mutation in the tyrosinase gene, making it necessary to examine the entire gene sequence to search for mutations. This is analogous to the situation with retinoblastoma, in which no hot-spot mutation in RB1 has been identified. Screening for OCA1 patients using rapid screening techniques such as heteroduplex/SSCP has proved useful.97 In terms of treatment, there is evidence that loading the diet with excess tyrosine may be an effective therapy for patients with OCA2.107,108
Aniridia
Molecular genetic studies of aniridia have provided a fascinating opportunity to examine the molecular mechanisms of ocular embryogenesis. Since the early demonstration of the 11p13 locus for autosomal-dominant aniridia and the association between sporadic aniridia and Wilms’ tumor, significant advances have been made in this field in recent years, leading to the identification of the human aniridia gene (PAX6). This congenital dominant eye disease is caused by haploinsufficient loss-of-function mutations of PAX6.109
Aniridia comprises a group of closely related panocular disorders with a common feature of iris hypoplasia (Fig. 55.3). Aniridia patients also can have a wide range of ocular findings, including cataract, foveal hypoplasia, nystagmus, corneal pannus, glaucoma, ectopia lentis, optic nerve hypoplasia, and strabismus. Aniridia can be genetically inherited either as an autosomal-dominant or an autosomal-recessive disorder (Gillespie syndrome).110,111 It is well known that the sporadic form of aniridia is associated with Wilms tumor, a pediatric nephroblastoma.112 The combination of sporadic aniridia and Wilms tumor is commonly referred to as WAGR syndrome. This may appear to be somewhat peculiar, since the familial form of the disease (autosomal-dominant or autosomal-recessive) is not associated with Wilms tumor.113 This apparent contrast is explained by the fact that PAX6 is located on chromosomal 11p13, in the vicinity of the gene for Wilms’ tumor.47,48,114,114a In the autosomal-dominant form of aniridia, the DNA alterations involved are often minor (point mutations or small deletions) and affect only the aniridia locus, sparing the Wilms tumor locus. In contrast, in the sporadic form of aniridia, there is often a large deletion of the gene segments involving the loci of both aniridia and Wilms’ tumor, resulting in the clinical presentation of both diseases.
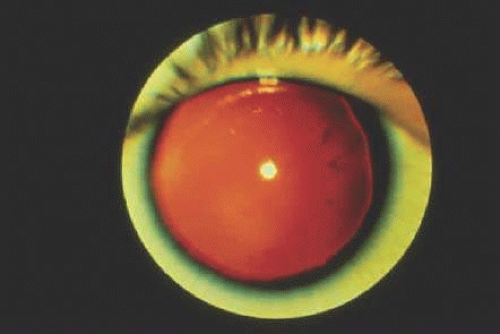 Figure 55.3. Aniridia. Decreased skin pigmentation and small residual amount of iris can be seen. |
PAX6 has been identified on chromosome 11p13.48,115,116,117 This gene contains 14 exons, is transcribed as a 2.7-kb mRNA, and encodes a 422-amino-acid protein. PAX6 may function as a transcriptional factor in regulating expressions of other genes in embryogenesis.116 Such an important role for PAX6 as the master control for gene expression in oculogenesis may explain the panocular manifestations in patients with aniridia. Aniridia and PAX6 have provided a valuable model system in the study of molecular mechanisms important in the embryogenesis of the eye.
Clinically, prenatal diagnosis can be carried out to detect PAX6 mutations in amniocytes.116 Such detection requires prior characterization of a PAX6 mutation in the family of the proband. In patients with the sporadic form of aniridia, it is important to detect deletions of chromosome 11p13 and the Wilms’ tumor locus because these patients may have a 50% risk of Wilms tumor development. Typically, karyotypes are examined and patients followed up with renal ultrasound. Molecular techniques are becoming available for direct detection of the number of copies of aniridia and Wilms’ tumor genes in patients with aniridia, thereby offering a genetic test for the predisposition to Wilms’ tumor.118,119,120,121
Axenfeld-Rieger Syndrome
Axenfeld-Rieger syndrome consists of bilateral congenital abnormalities of the anterior segment of the eye associated with abnormalities of the teeth, midface, and umbilicus. Ocular findings include prominent, anteriorly displaced Schwalbe line (posterior embryo toxon), peripheral iris strands extending to Schwalbe line, and iris thinning with atrophic holes (Fig. 55.4). Axenfeld-Rieger syndrome is typically inherited as an autosomal-dominant disorder,122 but 25% of cases are sporadic. There is no sex predilection. Cytogenetic studies have mapped the Axenfeld-Rieger locus to chromosome 4q25.123,124,125,126,127 Based on these linkage studies, a candidate gene located in this chromosomal region that encodes for epidermal growth factor (EGF)128,129,130,131,132 has been chosen. The EGF gene is considered an excellent candidate gene because EGF binds to the tooth, and antibodies to EGF inhibit dental and eye development.129,130,131 Antisense oligonucleotides to EGF have been shown to block tooth formation in vitro.133 In addition, Raymond et al130 and Jumblatt et al134 have shown that the corneal endothelium has EGF receptors and undergoes morphologic changes when exposed to EGF.134 However the hypothesis that EGF is the Axenfeld-Rieger gene is yet to be proved because no causative mutations in EGF have been identified to date in patients with Axenfeld-Rieger syndrome. Alternative candidate genes in this chromosome 4q25 region are still being sought.128 PITX2 and Dlx2 are two transcriptional markers observed during early tooth development. Mutations in PITX2 associated with Axenfeld-Rieger syndrome provided the first link of this transcription factor to tooth development. This mutation is associated with iris hypoplasia.135
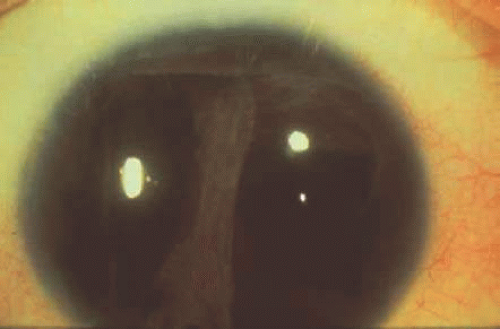 Figure 55.4. Rieger syndrome. Iris abnormality is prominent. |
Cataract
Hereditary cataract can be transmitted as an autosomal-dominant136 or X-linked trait and can be associated with other ocular findings (e.g., microphthalmia, microcornea) and systemic manifestations (e.g., dental anomalies).98,136,137,138,139 The lens opacity is often central in X-linked congenital cataract (Fig. 55.5). Using RFLP, the locus for X-linked congenital cataract has been mapped to chromosome Xp21.1-22.3.140,141,142 Many other chromosomal loci have been described for various other types of cataract, including Coppock-like cataract (chromosome 2q33-35),143 Mariner-type cataract (chromosome 16q22),144,145 anterior polar cataract (chromosome 2p25, 14q24),146 congenital total cataract (chromosome Xp),147,148 and juvenile onset hereditary cases have been reported to have a point mutation in the human γD crystalline gene.149
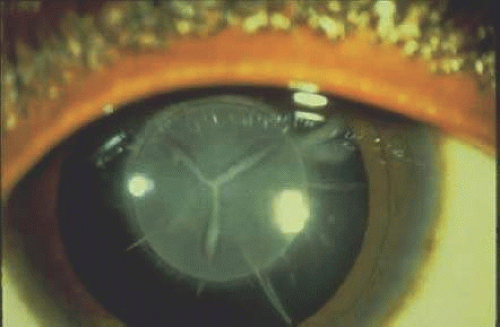 Figure 55.5. Congenital cataract characterized by the central location of the lens opacity. |
Choroideremia
The recent discovery of the molecular defect in choroideremia is an excellent example of the power of molecular studies in advancing our understanding of disease pathogenesis. Choroideremia is a hereditary, bilateral, and progressive X-linked retinal degeneration characterized by central blindness in affected males during early adulthood. These patients have hemeralopia, decreased vision, and visual field constriction due to atrophy of the choroid and the retinal pigment epithelium (Fig. 55.6). One notable characteristic of choroideremia in contrast with other retinal degenerations, such as X-linked retinitis pigmentosa, is that carrier females of choroideremia have a typical fundus appearance consisting of linear retinal pigmentation with punctate area of pigment epithelial atrophy.
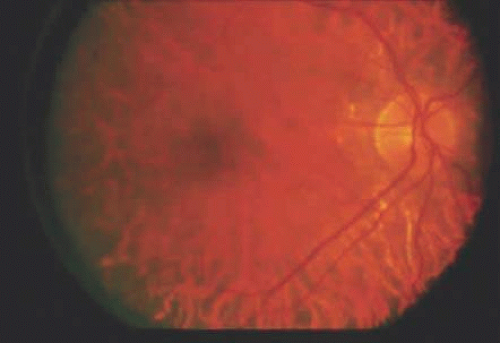 Figure 55.6. Choroideremia. Atrophy of retinal pigment epithelium is evident. |
The gene locus for choroideremia was mapped to the proximal region of Xq21.150,151,152,153 Through positional cloning, the achoroideremia gene has been identified and characterized in this region. Patients with choroideremia indeed have been found to have mutations in the putative disease gene.154,155,156,157,158,159 Up until 1993, however, very little was known concerning the biologic function of the choroideremia gene and its role in retinal degeneration. It was in the last 3 years that a significant breakthrough in this field was made by Seabra and colleagues,160,161 who reported that the choroideremia gene is in fact highly homologous to component A of rat Rab geranylgeranyl transferase, which belongs to a family of guanosine triphosphate-binding proteins. These proteins are believed to play important roles in the regulation of membrane transport and signal transduction. Furthermore, components of the rat Rab geranylgeranyl transferase have been found to be missing in patients with choroideremia.161 These small mutations involving the Rab escort protein-1 (REP-1) area ll nonsense, frameshift, or splice-site mutations with one possible exception, missense mutations have not been found. This comprises all the known mutations in the disease.162 The molecular basis of choroideremia appears to be due to a defect in the membrane transport of proteins with attendant abnormalities in signal transduction. In addition, these studies suggest the exciting possibility that the genes for a host of well-characterized membrane transport proteins can be considered candidate genes for a variety of other hereditary retinal degenerations.
Color Vision Defects
The molecular basis of human color perception has intrigued ophthalmologists for centuries. The remarkable advances achieved in this field in recent years are the result of modern molecular biologic studies, combined with insightful deductive reasoning and a close interaction between basic science research and clinical evaluation of patients.
Human color vision is mediated by three visual pigment proteins contained in cone photoreceptors: these pigments are sensitive to red (552 to 557 nm); green (530 nm); and blue (426 nm) light, respectively.163 Cone pigments function in bright light, in contrast to the rod photoreceptor pigment rhodopsin, which mediates vision in dim light. Visual pigment proteins are the main protein components of photoreceptor outer segments. Clinically, a person with normal color vision is called a trichromat. If one cone pigment is absent, the person is known as a dichromat. A protanopic dichromat is a person whose red cone pigment is missing, whereas a deuteranopic dichromat is a person whose green cone pigment is absent. Tritanopia, a rare disease, is the absence of the blue cone pigment. Patients lacking both the red and green pigments are known as having achromasy or blue-cone monochromasy. If all visual pigments are present but have altered absorption spectrums, the condition is termed anomalous trichromasia.
The phototransduction process starts with the capture of a photon by a visual pigment, leading to the change in conformation of 11-cis retinal chromophore contained in the visual pigment to a corresponding 11-trans structure. Activation of Gprotein and cyclic guanosine monophosphate (cGMP) phosphodiesterase causes an enzymatic cascade that eventually results in a change in the membrane potential of the photoreceptor and conduction of a visual signal.
To dissect the molecular basis of color vision, Nathans and co-workers164,165,166,167 reasoned in the early 1980s that if one assumes that the visual pigments have all evolved from a common ancestral gene, one can then clone the various human photopigments by starting with the bovine rhodopsin gene. Because the bovine rhodopsin protein sequence was known, Nathans and Hogness164 constructed a DNA probe with a nucleotide sequence corresponding to a small part of the bovine rhodopsin protein sequence. Using this DNA probe, they were able to obtain a complementary DNA (cDNA) clone and eventually sequence the entire bovine rhodopsin gene.
According to the plan of Nathans and Hogness,164 the bovine rhodopsin gene would serve as the anchoring molecule from which the human rhodopsin and all three human visual pigments were to be cloned. To isolate the human rhodopsin gene, they used bovine rhodopsin cDNA as a DNA probe and searched the human genome for homologous sequences. A human gene was identified and proved to be the human rhodopsin gene based on the fact that it shared a high degree of DNA sequence homology (89.7%) with bovine rhodopsin165,166,167 and that the amount of its mRNA in human retina corresponds well with previous observations.165 The human rhodopsin gene contains five exons and encodes for a protein with seven putative transmembrane hydrophobic domains. The gene is mapped to human chromosome 3q21.166
Nathans and co-workers next embarked on the cloning of all three human cone visual pigments. They reasoned that unlike the human rhodopsin gene, which shares a high degree of homology with bovine rhodopsin gene, the human cone pigments would share a lesser degree of DNA sequence homology. They therefore lowered the stringency requirement in the hybridization experiment. Again using the bovine rhodopsin cDNA as a probe, they obtained three human gene segments.167 The first segment showed 42% amino acid sequence homology with the bovine rhodopsin gene and was identified as the blue-cone pigment. The confirmation of the authenticity of the blue-cone pigment came from the observation that the amount of mRNA of the putative blue-cone pigment is about 1/150 of that of rhodopsin, consistent with the expectation based on the ratio of the number of blue cones to rods in the human retina167 and that the putative blue-cone pigment gene mapped to human chromosome 7, thus excluding it as being red or green cone pigment genes. The second and third human gene segments cloned were subsequently identified as the red and green cone pigments, respectively, based on the fact that (i) they also show 40% to 45% amino acid sequence homology with bovine rhodopsin and (ii) the ratio of the amount of mRNA of the putative red and green pigments to that of rhodopsin agrees with the ratio of red and green cones to rods (1/30).167 In addition, these two putative red and green pigment genes were mapped to human X chromosome as expected, and studies in red-green color-blind men further confirmed the identities of these two genes.
Red and green pigment genes are arranged in a tandem array on the human X chromosome, with the red pigment gene in the upstream position (5′ end). Each person has one red pigment gene, but a variable number (one, two, or three) of green pigment genes. The variation in the number and composition of these red and green pigment genes due to intragenic recombination events leads to variation in human color perception.166 Color vision defects are caused by deletions and fusions involving these genes.168 In an analysis of blue-cone monochromasy patients who showed X-linked absence of both red and green pigment, Nathans et al169 demonstrated inactivating DNA mutations in the red and green pigment genes. Furthermore, sequence changes in pigment opsins, which correspond to altered wavelengths at which the visual pigments absorb maximally,170,171,172,173,174,175,176 have been shown in patients. Other authors have found that defective color vision is caused by DNA mutations in the green pigment gene,177 and patients with tritanopia (autosomal-dominant disorder with lack of blue spectra sensitivity) have been shown to have mutations in the blue pigment gene.178 In addition, color vision defects as well as rhodopsin mutations have been found in patients with ADRP.49
Congenital Stationary Night Blindness
Clinically congenital stationary night blindness (CSNB) becomes manifest as night blindness from birth, normal visual field, and paradoxic pupillary responses. Patients may have a normal or abnormal fundus. The correct diagnosis of the disease is important because it is generally not a progressive disease. CSNB can be inherited as an autosomal-dominant, autosomal-recessive, or X-linked disease.179,180,181,182 Autosomal-recessive CSNB seems to be very rare.183 Myopia is often associated with autosomal-recessive and X-linked modes of inheritance. A CSNB patient typically shows variable reduction in rod function, but a functionally intact cone system. The disease is divided into two types, complete and incomplete, which correspond to an absent or reduced rod function, respectively.
The gene locus for X-linked CSNB has been mapped to chromosome Xp11.3.184,185,186,187 This chromosome region appears to play an important role in retinal function because, in addition to the CSNB gene locus, it contains two loci for X-linked retinitis pigmentosa (RP2188,189,190,191,192 and RP3192,193,194,195) and the locus for ocular albinism type 2 (OA2).98 Clinically, myopia and CSNB appear to be coinherited.187 This phenomenon could be due either to a closely linked myopia gene or to a secondary pleiotropic effect of CSNB. Further investigation of genes in this region of X chromosome may significantly advance our knowledge of X-linked retinal diseases.
Ectopia Lentis and Marfan Syndrome
Ectopia lentis is clinically characterized by a lens displayed away from its optical axis (Fig. 55.7). Ectopia lentis can be part of a hereditary disorder, such as Marfan syndrome, aniridia, homocystinuria, simple ectopia lentis, or congenital glaucoma.196 With regard to Marfan syndrome, significant advances have been made recently in our understanding of the molecular pathogenesis of the disease. Marfan syndrome is a dominantly inherited connective tissue disorder that is characterized by skeletal, cardiovascular, and ocular abnormalities caused by abnormalities of fibrillin metabolism. The Marfan disease-causing gene has been mapped to human chromosome 15.197,198,199,200,201 Because this chromosome region contains the fibrillin gene (FBN1),202 which is a major component of connective tissue, a candidate gene approach was taken to examine FBN1, and mutations have been identified in patients with Marfan syndrome.202,203,204,205,206 Genetic tests are becoming available for the molecular diagnosis of Marfan syndrome. This is significant because potentially life-threatening conditions are associated with this syndrome such as a high risk of aortic aneurysm and dissection.201
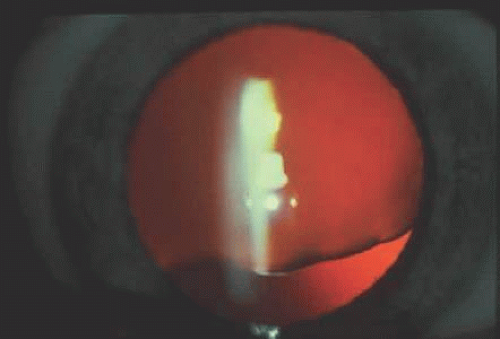 Figure 55.7. Ectopia lentis. This patient has Marfan syndrome with lens subluxed superiorly. |
Familial Exudative Vitreoretinopathy
FEVR was first described by Criswick and Schepens207 as an autosomal-dominant disease of the retina and vitreous. It is well known that the disease can be inherited both as an autosomal-dominant trait208,209 or as an X-linked recessive trait.210 FEVR patients typically present with bilateral incomplete vascularization of the peripheral retina, retinal exudation, neovascularization, and fibrovascular proliferation, which may lead to tractional or exudative retinal detachment.207,211–212a The funduscopic appearance is similar to retinopathy of prematurity, except that the patients do not have a history of premature birth and oxygen therapy. FEVR has a penetrance of nearly 100%, but its expressivity is variable.212,212a
Autosomal-dominant FEVR is genetically heterogeneous, but its principal locus, EVR1 is on chromosome 11q13-23.213 It is intriguing to note that this locus is the same as that found for autosomal-dominant neovascular inflammatory vitreoretinopathy,214 clinically a distinct disease characterized by neovascularization of the retina and iris, inflammation, pigmentary retinopathy, and cystoid macular edema. It remains to be determined whether FEVR and autosomal-dominant neovascular inflammatory vitreoretinopathy share the same gene defect and underlying molecular pathogenesis.19
The X-linked recessive form of FEVR has been mapped to two possible chromosomal loci, Xq21.3 or Xp11.4-11.3.215 Clinically, the X-linked recessive form produces an earlier onset of disease than the autosomal-dominant form.215 It is noteworthy that the Xp11.4 region also contains the Norrie disease gene, and patients with the X-linked form of FEVR have been found to have DNA mutations in the Norrie disease gene.56 It is anticipated that further elucidation of the molecular basis of these diseases will significantly facilitate clinical diagnosis and management (see Norrie Disease section later in chapter).
Glaucoma
Glaucoma is an important and prevalent ophthalmic disease. A large number of inheritable ophthalmic diseases are associated with glaucoma, such as aniridia (autosomal-dominant), Axenfeld Rieger syndrome (autosomal-dominant), neurofibromatosis (autosomal-dominant), and Lowe syndrome (X-linked recessive) to name just a few.216 In this section, we will confine ourselves to primary open-angle glaucoma and discuss the recent advances in understanding the molecular basis of the disease.
Primary open-angle glaucoma (POAG) has a strong hereditary tendency. Anyone who has taken care of POAG patients understands the importance in soliciting a relevant family history of glaucoma. Numerous studies have shown that the prevalence of POAG in first-degree relatives of patients (2.8% to 13.5%) is significantly higher than that in the general population (about 1% in the white population).217,218,219,220,221,222,223,224,225 Attempts have been made to identify genetic markers associated with POAG.226 These markers include blood group antigens,224,227,228,229,230,231 HLA antigen association,227,230,231 ability to taste phenylthiourea,232 and association with diabetes mellitus233 and myopia.234 These studies, however, have not revealed any specific association of these genetic markers with POAG. At present, POAG is believed to have a polygenic or multifactorial inheritance.
As is often the case in the history of human genetics, a rare disease may provide a valuable opportunity to gain insight into a mechanism of a related common disease process. We have seen this in the example of retinoblastoma, where the study of this rare childhood tumor has proved a model system of tumor-suppressor genes and human carcinogenesis. This is due to the fact that in rare diseases, the clinical phenotype and the molecular pathogenic process can often be clearly delineated. With regard to POAG, one strategy is to identify a certain subset of less prevalent but clinically distinctive forms of the disease. Through dissecting the molecular pathogenic process involved in these distinctive entities, one may gain valuable insight into the mechanisms of more common disease processes. The study of juvenile open-angle glaucoma (JOAG) provided just this sort of valuable opportunity. It has been known for some time that JOAG is an autosomal-dominant trait.235,236,237,238,239 In 1993, Sheffield and associates240 reported genetic linkage of one form of familial open-angle glaucoma to chromosome 1q21-q31. Other groups have reported continued effort in cloning the causative gene in this chromosome region.25,241,242
The future identification of gene defects in JOAG and POAG will no doubt have a dramatic effect on the clinical management of glaucoma patients. Appropriate molecular diagnostic tests can be established. This is particularly important for glaucoma: Because of its insidious manner of onset and the relatively asymptomatic nature of the disease in the early stages, patients often present for medical attention only when the disease is significantly advanced and irreversible damage to the optic nerve has occurred. Molecular diagnostic tests will enable the clinician to recognize individuals who are predisposed to or are at an early stage of the disease. Knowledge of the gene defects and their association with the severity of clinical presentation will have important prognostic value. Furthermore, recognition of key molecular events in the disease process will help create new forms of therapy for this condition.
Gyrate Atrophy
Gyrate atrophy is an autosomal-recessive disease. Clinical findings of this disease include multiple sharply defined areas of chorioretinal atrophy separated from each other by thin margins of pigment (Fig. 55.8). The lesions typically begin in the midperiphery in childhood and then progress and coalesce to involve the entire fundus, sparing the fovea until later in the disease process, usually in midlife. Ornithine levels are markedly elevated in all body fluids.
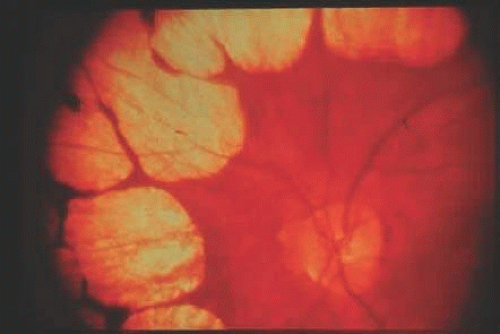 Figure 55.8. Gyrate atrophy. There are multiple sharply defined areas of chorioretinal atrophy separated from each other by thin margin of pigment. |
Gyrate atrophy genetics is one of the few examples in ophthalmology of classical forward genetics: that is, the protein defect of the disease (ornithine aminotransferase [OAT]) was known before the chromosomal localization of the causative genes.243,244,245 An example of a nonophthalmic disease in which classical forward genetics was employed is sickle cell anemia246: the protein (hemoglobin) defect was known before the chromosomal localization of the gene. In the last few decades, however, reverse genetics has become the dominant strategy in modern gene cloning, as demonstrated in the case of CF.1
The gene encoding for OAT has been mapped to chromosome 10q26.247,248,249,250 The OAT gene consists of 11 exons and spans 21 kb of genomic DNA and 2.2 kb of mRNA.251 Several mutations in this gene have been identified in patients with gyrate atrophy.245,250,251,252,253,254,255,256,257,258,259,260,261,262,263,264,265,265a,265b There are two types of gyrate atrophy: vitamin B6 (pyridoxine) responsive and vitamin B6 nonresponsive. The first group typically responds to the administration of vitamin B6, which reduces the serum ornithine level. Genetic heterogenicity of gyrate atrophy has been suggested by the demonstration that administration of pyridoxine to increase levels of pyridoxal phosphate, a cofactor of OATase, reduces hyperornithinemia in a subset of patients. These data firmly establish that both pyridoxine responsive and nonresponsive forms result from mutations in the OATase gene.266 It is still not known why OAT gene mutations preferentially affect ocular tissue, since OAT gene is expressed systemically.
Leber Hereditary Optic Neuropathy
The molecular genetics of Leber hereditary optic neuropathy has become a prototypic example of mitochondrial inheritance. With the identification of the underlying molecular defect, this disease has provided a unique opportunity for examining the relationship between DNA mutations and clinical presentation of the disease.
Originally described in 1871 by Leber,267 Leber hereditary optic neuropathy is characterized by rapidly progressive and painless visual loss in first one eye and then, within days to months, the other eye. The disease usually occurs in young men 15 to 30 years of age. Clinically there is mild swelling of the optic disc progressing for a period of weeks to optic atrophy. Small telangiectatic blood vessels can be seen near the optic disc (Fig. 55.9) that do not leak on intravenous fluorescein angiography. Visual acuity is often 20/200 to counting fingers, and visual field examination typically demonstrates a cecocentral visual field defect. It has been known since Leber original description of the disease that Leber hereditary optic neuropathy has a unique maternal transmission pattern: the genetic defect appears to be carried and transmitted from mothers to their children (usually sons), and affected fathers do not transmit the disease gene to their offspring. It was thus speculated that mutations in the mitochondria might be responsible for this disease because mitochondria are passed exclusively from mothers to offspring, not from fathers.
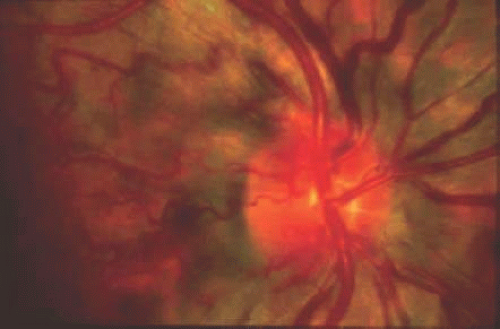 Figure 55.9. Leber’s hereditary optic neuropathy. Disc hyperemia and dilated and telangiectatic vessels can be seen. |
The mitochondrial inheritance was confirmed in 1988 by Wallace et al who identified a mitochondrial DNA replacement mutation at nucleotide position 11778. The first DNA mutation was identified in mitochondrial DNA in patients with Leber hereditary optic neuropathy.268,269,270,271,272,273,274,275,276 These mutations can be categorized into two types: primary mutations, believed to be disease-causing DNA alterations; and secondary mutations, which act synergistically with primary mutations in modulating the clinical severity of the disease. Among the primary major mutations identified, point mutations in mitochondrial DNA nucleotide positions 11778, 3460, and 14484 account for 50%, 30%, and 10% of the disease, respectively, as seen clinically.269,270,271,272,273,274,275,276 The most prevalent mutation (at nucleotide position 11778) is a transition mutation that converts G to A, resulting in the replacement of a conserved amino acid arginine to a histidine. The 11778 mutation is associated with poor visual prognosis.277,278 Clinically, these mitochondrial mutations can be classified as high-risk (class I, or primary), intermediate-risk (class I/II, or mixed primary and secondary), or low-risk (class II, or secondary) mutations.
It is intriguing to examine the molecular pathogenesis of Leber hereditary optic neuropathy, and several observations appear to be important in correlating DNA mutations with clinical disease:
Mitochondria are vital intracellular organelles that supply energy for cellular metabolism through the electron transport chain system. Mitochondrial DNA mutations identified in Leber hereditary optic neuropathy all appear to occur in genes involved in the mitochondrial metabolic process. For example, the 11778 mutation occurs in the gene ND4, which encodes subunit 4 of NADH dehydrogenase of the respiratory chain complex I, an enzyme necessary for the synthesis of adenosine triphosphate. Experimental evidence suggests that mutations such as ND4/11778 may affect the rate of NADH-dependent oxidative processes.279,280 It appears that mutations such as the 11778 mutation that occur at critical positions of the mitochondrial genome give rise to more severe phenotypes.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


