Molecular Biology and Genetics of the Retina
John M. Nickerson
J. Fielding Hejtmancik
The reference material presented in this chapter provides the resident and clinical ophthalmologist with an adequate knowledge base to follow the exciting advances in the field of molecular biology of the retina and other structures of of the eye. Many of these advances are now the definitive genetic tests for certain ophthalmic diseases. The clinical practitioner needs to be not only familia with these tests But also aware of some caveats associated with them. Thus, the function of this chapter is to provide a primer for the fundamentals of molecular biology, examples of the application of molecular biology to fundamental research on the basics of the retina and visual system, applications to specific retinal diseases and degenerations, and finally a discussion of the powerful techniques underlying the world of genetic linkage tests. This chapter is not intended to replace a survey course in molecular biology or to provide detailed protocols for carrying out these techniques (several excellent texts1,2 and laboratory manuals3,4 provide thorough treatment of these topics). Also, this chapter is not meant to be encyclopedic on all the current topics of retinal molecular biology. In the following section, we only touch on a comparative handful of these, beginning with a few definitions.
OVERVIEW OF TERMS AND TECHNIQUES
MOLECULAR BIOLOGY
Molecular biology is the detailed study in detail of polymers of nucleic acids (DNA and RNA) that encode gene products, largely proteins. It also is the study of the control and regulation of gene expression. “Molecular biology is a discipline, a level of analysis, a kit of tools—which is to say, it is unified by style as much as by content. The style is unmistakable. The style is bold; it is simplifying; it is unsparing; often it is extremely competitive. The style is also, sometimes, subtle and sophisticated.”5
DNA
DNA is the fundamental genetic material that encodes genes. In almost all forms of life, DNA is replicated and passed from one generation to the next. Information is stored in DNA by the sequence of the bases in the molecule: adenine (A), guanine (G), cytosine (C), and thymine (T). By convention the sequences from left to right are the 5′ to 3′ orientation of the sense strand of the DNA molecule, where “5’” is the fifth position on the deoxyribose and the “3’” refers to the third carbon atom of deoxyribose. Information about a gene product is stored in groups of three bases. The genetic code decodes the triplets of bases, translating the DNA sequence through its messenger RNA (mRNA), an RNA copy of the DNA sequence, into an amino acid sequence. The amino acid sequence folds into a three-dimensional structure, forming a protein that is capable of some biologic function. For the most part proteins, rather than DNA or RNA, constitute the physiologic cellular machinery. Recent developments suggest that small RNAs play important roles in metabolism and that small RNAs may be used therapeutically to interfere with the expression of some genes.6
RESTRICTION ENZYMES
The discovery of restriction enzymes by microbiologists studying Escherichia coli and other bacteria provided a foundation for modern molecular biology. These enzymes, which are found in a variety of prokaryotes, cut the DNA double helix at specific base sequences, or sites. These sites are short but specific DNA sequences, usually four to eight bases in length. The natural function of a restriction enzyme is to degrade the DNA of invading viruses in order to protect the bacterium from infection. For example, the restriction enzyme EcoRI recognizes the sequence GAATTC in DNA and cleaves the DNA between the G and the first A. Over 500 different restriction enzymes, each recognizing a different DNA sequence, are now commercially available. They usually recognize sequences that are palindromic (reading the same forward and backward).
When these enzymes cut the DNA strands in the middle of their recognition sequence, blunt ends are formed. When the strand scissions are offset, overlapping single-stranded ends of two or four bases are created. These “sticky ends” can occur with an overhang of the 3′ strand (e.g., as produced by the restriction enzyme, Pst I) or with the 5′ strand (e.g., as with EcoRI). Because any sticky ends created by digestion with the same restriction enzyme can reanneal readily under the proper conditions, these enzymes provide a powerful tool for inserting foreign DNA fragments into many vectors or other constructions. This allows virtually all sequences from the human genome to be cloned (inserted into plasmid or bacteriophage vectors and replicated in bacteria), so that large amounts can be isolated in pure form.
One way in which a bacterium protects its own DNA from digestion with its own restriction endonuclease is with a methylase that recognizes and adds a methyl group to the given sequence. This sequence, once methylated, is no longer a target for some restriction endonucleases. Other restriction endonucleases are not inhibited by methylation, and these can recognize exactly the same sequences as the homologs that are inhibited. Two restriction enzymes that recognize the same base sequence but are isolated from different organisms are called isoschizomers. Just as isoschizomers may or may not react similarly to methylation, they may not cut the DNA strands at the identical site within the recognition sequence, and thus they may not produce identical ends.
Although these differences are of more central concern to cloning and rearranging DNA fragments, they also can be important in gene mapping. For example, the use of a methylation-sensitive restriction endonuclease for analysis of restriction fragment length polymorphisms (RFLPs) might suggest genetic (DNA sequence) polymorphism when none exists. Methylation plays a role in gene expression in higher eukaryotes, and the use of pairs of isoschizomers, one of which is inhibited by methylation, is a means of assessing gene inactivation. Usually a gene that is not expressed in a given tissue is methylated, whereas the same gene is not methylated in a tissue where the gene’s expression is needed.
RECOMBINANT TECHNOLOGY
Recombinant DNA technology is the set of techniques and tools used by molecular biologists to manipulate DNA molecules. Recombinant DNA technology allows the preparation of large amounts of a pure and homogeneous DNA sequence. Figure 1 illustrates the fundamentals of the technology with a procedure called subcloning. This procedure begins with a DNA sequence of interest, for example, part of a gene or cDNA that will be “inserted” into a second, usually larger, piece of DNA, called a “vector,” which is used to propagate the gene fragment. Vectors usually are derived from bacterial viruses (bacteriophages) or bacterial plasmids, small double-stranded DNA circles commonly found in bacteria that replicate autonomously from the bacterial DNA. The vectors usually have been manipulated to delete parts of the parental plasmid or virus and frequently have had other genes added to them depending on their specific application. To subclone the gene fragment into a different vector, some sequence information or knowledge of the positions of restriction enzyme recognition sites is required. The sequences of the vectors are usually known. Using a desktop computer and DNA sequence analysis software, restriction enzyme sites in sequences can be identified readily. The choice of the enzymes depends on the availability of compatible ends that border the insert to be subcloned, the absence of extra internal sites in the insert, and compatible sites in the vector. Once the gene fragment has been cleaved with a restriction enzyme, the DNA fragments of DNA can be purified by any of several techniques, including electrophoresis, differential precipitation, and chromatography. The purified DNA fragment can be introduced into a specialized vector that has a compatible restriction enzyme site in an appropriate location. The two DNA molecules are mixed in roughly equimolar amounts, and the enzyme T4 DNA ligase is added. This enzyme will join covalently the 5′ phosphate from the end of one DNA molecule to the 3′ hydroxyl of another DNA fragment. When both ligations (one from each end of the two molecules) have been completed, we will have created a circular DNA molecule bearing a copy of the vector and the insert. By judicious choice of conditions in the previous ligation reaction, the fraction of aberrant molecules can be minimized but not totally eliminated. Important examples of these unwanted circular molecules include a single copy of the vector with two or more inserts and others that have no insert.
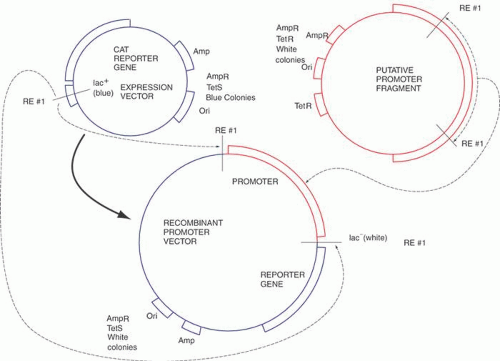 Fig. 1. Subcloning of a DNA fragment, in this case containing a putative promoter fragment, from one clone into another vector. Judicious selection of an appropriate restriction enzyme yields a recombinant plasmid containing the inserted promoter fragment in the recombinant plasmid. Drug resistance markers are located on each of the plasmids, allowing selection of the appropriate plasmid after bacterial transformation. The plasmids contain enough markers so that it is easy to discriminate among the three plasmids shown in the figure. The expression vector contains the β-lactamase gene, which allows bacteria containing this plasmid to grow in the presence of ampicillin. This plasmid also contains the gene for β-galactosidase. In the presence of a substrate called X-gal, colonies containing this gene will turn blue. The restriction enzyme (RE) site for RE 2.1 is within this gene, and when a fragment is successfully cloned into this site, the gene is interrupted and is no longer capable of functioning to convert the substrate, X-gal, into the blue product. Thus, colonies with the insert in the expression vector will be white. Bacterial colonies which are TetR and AmpR (tetracycline resistant and ampicillin resistant, respectively) result from the original plasmid containing the promoter fragment. Neither of the other plasmids (the expression vector or the recombinant vector) contain the TetR gene, and they will be TetS (sensitive to tetracycline), so colonies containing these plasmids will fail to grow on plates containing tetracycline. To select the appropriate colonies, we plate the initial transformants on Amp plates containing X-gal. One day later we will pick the white colonies. We will replate them on Amp plates in an ordered pattern and on Tet plates in the same ordered pattern. Those that are AmpR, white, and TetS will be the colonies that contain the appropriate plasmid. We will verify the legitimacy of the plasmid by preparing a small amount of the plasmid from these colonies and digest the plasmid with RE 2).1, the restriction enzyme that we used to make the construct. Valid constructs should give us the two identically sized fragments that we used to make the recombinant plasmid. |
The next step, bacterial transformation, introduces the vector-insert recombinant plasmid into bacterial cells. This can be done by chemical treatment or by electroporation, both of which open small pores in the bacterial plasma membrane, allowing the DNA to enter the cell. Once in the bacterium, the plasmid replicates independently of the host cell DNA and continues to do so long after the cell stops growing. Depending on the particular plasmid, each bacterial cell might contain several to hundreds of copies of the plasmid. Usually the vector DNA contains an antibiotic resistance gene, such as β-lactamase, that allows a transformed bacterial cell harboring the recombinant plasmid to grow in media containing an antibiotic, ampicillin in this case. By plating the bacteria on Petri dishes containing a rich medium including ampicillin, only resistant bacteria (those carrying the plasmid) will grow enough to form appreciable colonies in a day or so. The transformation events are rare enough so that individual colonies can be chosen, selected with a sterile loop or toothpick, and replated. Thus, in only 1 to 2 days, a single bacterial colony can be purified to homogeneity that bears a single pure plasmid. A milligram of the plasmid can be purified readily from a 1-liter culture of the isolated bacterium. This quantity is usually sufficient to last for 1 year.
SOUTHERN BLOT ANALYISIS
Digestion of a small plasmid (e.g., pBR322, which is 4,362 bp in size) with a restriction endonuclease produces a small number of fragments of discrete size. By subjecting the digested DNA to agarose gel electrophoresis, these fragments can be separated by their sizes and then detected visually with ethidium bromide staining. Ethidium bromide binds to DNA, and when bound, it fluoresces quite intensely. In electrophoresis, the electric field moves DNA fragments through the meshwork of agarose fibers. Because the agarose matrix impedes larger DNAs more than smaller fragments, small DNAs migrate faster than the larger fragments. After electrophoresis and staining, the resulting DNA fragments appear as an array of bands on the agarose gel, with the larger fragments near the starting point and smaller fragments progressively nearer the anodal end of the gel. The set of bands is a fingerprint characteristic of the plasmid that was digested with the one restriction enzyme. It is easy to compare different plasmids digested with the same enzyme by running the digests on adjacent lanes of an agarose gel.
To learn whether a DNA sample, e.g., the insert of a plasmid, contains some or all of a gene or to learn whether the insert is a faithful representation of the gene in a human being, it is often necessary to evaluate human DNA on an agarose gel. When DNA isolated from humans or other higher eukaryotes is digested with a restriction enzyme, the result appears as a broad smear from top to bottom of the gel. This is because the human genome contains 3 × 109 base pairs (the number of unique nucleotides present in a haploid set of human chromosomes). For a restriction endonuclease recognizing a sequence of six nucleotides, assuming recognition sites are randomly distributed approximately once every four kilobase (kb) pairs in the genome, resulting in roughly 7.5 × 105 different DNA fragments. The individual fragments lie so close together that they appear as a smear on an ethidium bromide–stained gel. Clearly, if an individual fragment is to be identified, a means other than ethidium bromide staining is required to locate the band on the gel.
The double-stranded nature of DNA, with Watson-Crick base pairing between the two strands containing complementary sequences, provides an ideal means to identify specific DNA fragments. After being run on an agarose gel, the DNA fragments are denatured to separate the complementary strands and then transferred from the agarose gel onto a support membrane, either nitrocellulose or more commonly nylon (Fig. 2). The transfer can occur by capillary action (the classic Southern blot), vacuum, pressure, or electric current. While the initial binding of the DNA to the membrane is reversible (unless the transfer is performed in alkaline buffer), the fragments are covalently attached by baking, ultraviolet irradiation, or sometimes merely drying.7 The DNA fragments are immobilized on the membrane in a denatured state and in a position precisely corresponding to their locations on the agarose gel.
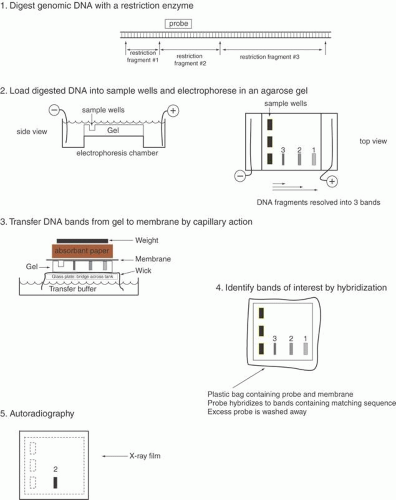 Fig. 2. Southern blot analysis. To identify a specific genomic DNA fragment among the hundreds of thousands of restriction enzyme fragments in the human genome, we use the Southern blot technique.7 This approach consists of several steps illustrated above. Genomic DNA is prepared and digested with any of several restriction enzymes. The DNA fragments are separated by size on agarose gel electrophoresis. The smaller fragments migrate faster through the agarose fiber meshwork. The fragments of interest can be identified by hybridization with a labeled probe. The DNA bands are first transferred out of the agarose onto a nitrocellulose or nylon membrane by capillary action. The DNA fragments can be permanently attached to the membrane by heat or ultraviolet light treatment. A radiolabeled DNA probe (e.g., a denatured cDNA) in single-stranded form can anneal by base pairing to its counterpart on the membrane. Excess probe DNA is washed off the filter, and by autoradiography of the filter we can visualize the specific DNA band or bands matching the probe. The Southern blot procedure allows us to detect and measure the size of one or a few DNA fragments out of the millions of different genomic DNA fragments produced by digestion with a given restriction enzyme. |
Specific DNA fragments are identified by annealing with a probe that has been labeled with radioactive phosphorus or fluorescent nucleotides. The probe is usually a piece of cloned DNA isolated from its vector. The probe is denatured into its two strands just before application to the membrane. This allows each strand of the probe to anneal to the denatured DNAs bound to the membrane. Radioactive nucleotides are incorporated into probes in several ways. Nick translation involves nicking one strand of the DNA double helix with DNase I and then elongating from this point by E. coli DNA polymerase I replacing one strand with radioactive nucleotides. In random priming, a mixture of many different oligonucleotides, usually hexamers, is annealed to denatured DNA strands to serve as a primer for elongation of the templates. This technique has the advantage of giving very high specific activities (usually greater than 109 cpm (counts per million) per μg with 32P-labeled nucleotides) and can be used to label DNA fragments in low melting point agarose directly after excision from an electrophoretic gel. Various alternative means to label probes, including production of a single-stranded probe with M13 phage or riboprobes (labeled RNA probes), are useful in special circumstances. When oligonucleotides are used as probes, they are simply end-labeled with polynucleotide kinase, which transfers the Γ-phosphate (usually bearing radioactive 32P) from adenosine triphosphate (ATP) to a 5′ hydroxyl group in DNA.
The probe, if initially double stranded, is denatured thermally or by adjustment to an alkaline pH before hybridization. It is added to a suitable hybridization medium and allowed to anneal to the target DNA immobilized on the blot. Annealing or hybridization takes place best at approximately 5° C below the melting temperature (Tm) of the probe-target complex. The Tm can be influenced by the hybridization solution in several ways: it will increase in proportion to the logarithm of the ionic strength of the solution, and it will decrease in the presence of formamide or other denaturant. The Tm is influenced by the length of the probe and the percentage of bases that are G and C. G:C base pairing is more stable than A:T base pairing. A general formula for the melting temperature of a hybrid is:
Tm = 81.5° C – 16.6(log10[Na+]) + 0.41(% G + C)– 0.63(% formamide) – (600 / probe length)
where [Na+] is the sodium ion concentration in molar dimension, and probe length is in base pairs. In addition, the Tm of a hybrid is decreased by 1 × to 1.5 × for each percent of mismatch between the probe and its target sequence. Hybrids formed by RNA are slightly more stable, so an RNA-DNA hybrid might melt at a temperature as much as 10° C above the corresponding DNA-DNA hybrid. Finally, short oligonucleotides follow a different rule:
Tm = 2(A + T) + 4(G + C)
where each letter represents the number of that particular base in the oligonucleotide. For example, the Tm of a 20-mer with four A’s, six G’s, five C’s, and five T’s is 62° C. There are several protocols for hybridization in common use, each of which works well if used appropriately. These protocols have several ingredients in common. Ionic strength (Na+ concentration, other monovalent cations) and denaturants such as formamide influence the stability of specific hybrids. A second group of components including detergents (especially sodium dodecyl sulfate) and charged species (such as bovine serum albumin, polyvinyl pyrolidone, and ficoll in Denhardt’s solution) tend to decrease nonspecific binding of the probe to the membrane, which is usually dependent on charge phenomena. Finally, components such as herring sperm DNA are designed to decrease nonspecific binding of repetitive elements in the probe to target DNA. Probes that contain repetitive elements also can be preannealed for several hours to a low Cot value with sheared human DNA so that these sites in the probe are “protected” and cannot recognize repetitive DNA on the membrane immobilized target DNA. This preannealing step blocks the repetitive elements in the probe so that only unique sequences of the probe are available to hybridize to DNA bound on the membrane. This decreases nonspecific binding when the probe is used for hybridization. Dextran can be included to increase the rate of hybridization. Usually the probe is incubated with a filter overnight to assure the specific hybridization of the probe to the filter-bound DNA reaches equilibrium.
After hybridization of the probe to filter-bound DNAs, excess nonspecifically bound probe is rinsed off by a series of incubations in large volumes of buffer. Initially, nonstringent washes are performed. These buffers are usually approximately 300 mM salt and near room temperature. These preliminary washes remove most excess radioactive probe and are followed by stringent washes that are conducted at elevated temperatures, near the Tm of the probe. The buffer for a stringent wash contains about 15 mM salt, and the wash temperature is set at 52° to 68° C for 20 minutes. There are two to four changes of the stringent buffer. Under these conditions only tightly and specifically bound probe remains attached to the filter. Afterward, the membrane with the specifically bound probe is wrapped in Saran Wrap or sealed in a plastic pouch, placed in a light-tight cassette on a piece of x-ray film (autoradiography), and left to expose the film for 1 to 2 days. Usually two intensifying screens are placed in the cassette to shorten exposure time. To reduce reciprocity failure of the film, the exposure is normally carried out at -80° C. This low temperature reduces thermal decay of the first of the two interactions required for a latent image to form on the film. The image on the developed film identifies the hybridizing DNA bands. The comparison of cloned gene fragments with authentic human DNA can validate the cloning process and show whether the entire gene was cloned. Also, Southern blot analysis7 can show differences among individuals at particular genetic loci, which can be used in linkage analysis, as discussed later, or in the identification of mutations that cause inherited diseases.
Besides the classic agarose gel electrophoresis method, other techniques for separating large fragments of DNA have been devised. Pulsed-field and field inversion gel electrophoresis allow separation of extremely large DNA fragments. These electrophoretic techniques are discussed later. Once these tests are run, a similar Southern blot technique can be used to identify a specific large DNA fragment. These combined approaches have been useful in analyzing bacterial artificial chromosome (BACs) and yeast artificial chromosome (YACs) clones, both of which contain large DNA inserts.
cDNA LIBRARIES, GENOMIC LIBRARIES, AND LIBRARY SCREENING
Within a given tissue, only a small fraction of the genes of the whole genome is expressed. In the retina, opsin mRNA makes up about 2% of the total mRNA, and interphotoreceptor retinoid-binding protein (IRBP) mRNA accounts for approximately 0.1%. Perhaps as few as 10,000 different mRNAs are expressed in the retina, whereas there are approximately 35,000 genes expressed in the body at different times. Ideally, cDNA libraries are complete collections of DNA copies of these mRNAs that have been cloned in an appropriate vector. The process of constructing such a library is shown in Figure 3. The key step is the conversion of RNA to DNA by the enzyme reverse transcriptase. Reverse transcriptase requires a double-stranded region to initiate synthesis of DNA. A synthetic DNA oligonucleotide can serve as a primer for the reverse transcriptase. This primer anneals to the mRNA providing the necessary double-stranded region. In many cases the primer used is oligo(dT). This primer can base-pair to a poly(A) stretch at the 3′ end of most mRNAs, and poly(A):oligo(dT) provides the double-stranded region that reverse transcriptase needs. The initial DNA copy of the RNA is converted to double-stranded DNA by the Klenow fragment of DNA polymerase I. Several alternatives can be used to modify the ends of the cDNA to make it compatible with cloning vectors. Here we illustrate the use of tailing reactions with terminal deoxynucleotidyl transferase. The remaining reactions are similar to the subcloning strategy shown in Figure 1. The main difference is that we are now treating a mixture of thousands of different cDNAs in the same way that we previously treated the single homogeneous DNA insert. Because of the greatly increased number of different inserts, the steps of ligation into the vector and transformation must be efficient to ensure that no mRNA molecule or its corresponding cDNA is lost from the library.
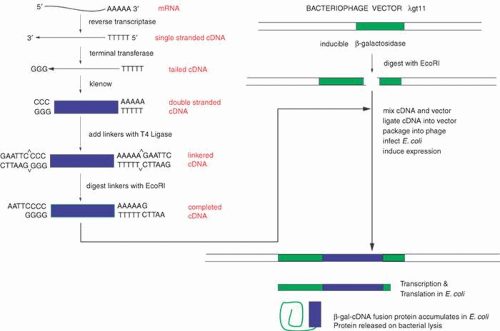 Fig. 3. Construction of a cDNA Library. To clone a large group of cDNAs from a given tissue, such as the retina, we begin with a few milligrams of RNA. Only approximately 0.5% to 5% of the total RNA represents mRNA; the bulk of the RNA is ribosomal and tRNA. Reverse transcriptase copies the mRNA into single-stranded DNA, complementary DNA (cDNA). This is converted to double-stranded DNA by the Klenow fragment of DNA polymerase I. Linkers are ligated to the ends of the cDNA. When digested with a restriction enzyme, such as EcoRI, these give sticky ends compatible with similarly digested vector. The thousands of different cDNAs are mixed with an excess of the digested vector, and T4 ligase is added. In suitable conditions only one cDNA will be ligated into each vector molecule. The recombinant vector, λgt11 with a cDNA, is packaged to reconstitute viable bacteriophage and can be grown on agar plates seeded with enough Escherichia coli to form a uniform lawn of cells over the surface of the plate. Because of the design of the vector, the infected E. coli will produce some protein derived from the cDNA. This protein also contains some amino acid sequences from the vector, and the protein is a hybrid of bacterial galactosidase and the polypeptide from the cDNA. This is called a fusion protein. The group of bacteriophage resulting from the insertion of the individual cDNAs into the vector molecules is called a cDNA library. |
Another way to minimize these losses is to make very large cDNA libraries, with 107 or more clones. Such a large library is redundant, containing many copies of the same mRNAs, but these libraries should be relatively complete, hopefully with greater than 95% of all possible mRNAs represented in them. The ligation of the cDNA to the vector usually allows for only one cDNA per vector molecule. Thus, colony isolation and purification produces one pure cDNA from one pure bacterial colony or phage plaque. Although the clones in most cDNA libraries directly reflect the frequencies of various mRNAs in the tissue of origin, special techniques allow the construction of libraries containing sequences unique to a specific tissue or developmental stage (subtraction libraries) or with roughly equal representation of all mRNAs found in a tissue (normalized libraries).
To find a cDNA clone among the thousands or millions of other clones represented in a library requires ingenuity and hard work. The first clone is always the hardest to find; after that it can be used as a hybridization probe to obtain longer clones or to walk to either end of the mRNA. One classic method to identify a cDNA clone coding for a specific protein is illustrated in Figure 4. Antibodies produced against the protein for which a cDNA is sought may bind to antigens that are produced on a protein that is expressed in E. coli as a fusion protein. The fusion protein is encoded by the cDNA that has been inserted into an expression vector, here λgt11. (Parts of the fusion protein are also encoded by the vector.) The antibody-antigen reaction can be detected by a color reaction, which generates a spot on a replica of the surface of a Petri dish covered with plaques. The location of the recombinant phage that contains the cDNA we want to isolate is the area where a colored spot coincides with a plaque. Multiple rounds of plaque purification ensure that the clone is homogeneous. Other schemes and strategies that also yield the desired clones including the following.
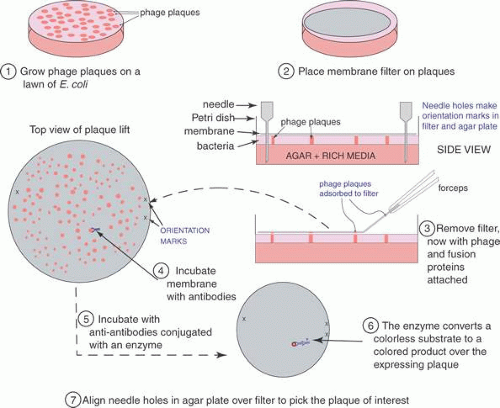 Fig. 4. The cDNA library screening process. cDNA libraries contain millions of different bacteriophage particles. To find the one bacteriophage out of the millions that contain the cDNA of interest requires an efficient and rapid technique to simultaneously analyze (nondestructively) all of these different cloned molecules. The procedure begins with the library plated on petri dishes. Typically, up to 500,000 phages might be grown on each 10-cm plate. A nitrocellulose filter is placed on top of the plate. This adsorbs some of the material on the surface of the dish, including bacteria, phage, and debris from lysed bacteria. This adsorbed material also includes some of the fusion protein expressed by bacterial cells before lysis kills an infected cell. This fusion protein leaks out of the bacterial cell, and some of the fusion protein will absorb onto the nitrocellulose filter where the plaque is located. For orientation purposes, marks are made in the filter and the petri dish with a syringe needle. These are needed to align the processed filter and the master plate to pick out the phage plaque with the positive signal on the filter. To detect the positive plaque, we incubate antibodies raised against the protein for which we want the cDNA clone.181,182 These antibodies bind to the fusion protein containing antigenic determinants common to it and the protein. Excess antibodies are rinsed off, leaving only the antibodies bound to the specific fusion protein. Again, these are located on the filter positioned directly over the spot where the plaque producing the fusion protein is on the plate. Second antibodies (anti-antibodies) conjugated with an enzyme such as alkaline phosphatase are incubated with the filter. After rinsing off excess antibodies, a substrate, which produces a colored product, is added, and a colored spot develops on the filter corresponding to the plaque producing the fusion protein. We align the filter and the plate, and with a glass pipet excise the plaque(s) corresponding to the signal. The phage from this plug of agar are replated, and the procedure is repeated until the phage are 100% positive for the antibody reaction, indicating that the bacteriophage is pure. Each of the several methods described previously has been used successfully, but the most effective has been the first, the use of antibodies and an expression cDNA library. |
Expression of pooled cDNA clones followed by functional biologic tests of the expressed proteins. The pools are then broken down into smaller and smaller pools until a unique clone is identified. This strategy is similar to the one discussed previously, except that in place of an antigenic reaction, a biologic function of the expressed protein is used in its place.
The use of a degenerate oligonucleotide probe. By reversing the genetic code, a set of potential oligonucleotides can be obtained that might encode the protein for which we seek the cDNA. This strategy depends on the prior determination of at least some amino acid sequence of the protein that we want.
Hybrid—selection. Groups of cDNA clones are used to select specific mRNAs from the total mRNA (the mixture of thousands of different mRNAs isolated from a tissue such as the retina) by hybridization of the mRNA to pools of cDNAs bound to a nitrocellulose filter. Specific mRNAs bind to the filter-bound cDNA clones, whereas the bulk of the mRNAs wash through; the specifically bound mRNAs can then be eluted later in purified form. These selected mRNAs can be translated to identify which proteins they produce. Similar strategies include the production of mRNA from individual clones (in suitable vectors) with T3 or T7 RNA polymerase and 7-methylguanosine Cap analogues. The resulting RNAs can be translated and analyzed.
Subtraction screening. When there is little information available about the properties of the protein, it may be necessary to resort to this strategy, which has been applied very successfully, especially if the expression of a gene can be identified in a first tissue and shown to be absent from a second similar tissue. RNA from the first tissue is hybridized to an excess of cDNA from the second. The RNA that does not hybridize to the cDNAs is separated from the bound RNA and should contain a greatly enriched preparation of the mRNA that we want to clone. Several variations of this strategy have been used successfully; for example, the mouse rd1 (retinal degeneration 1) gene was first isolated by this strategy. Wild-type retinas contain photoreceptors, but rd1 retinas selectively lose theirs. Bowes and associates applied subtraction strategies to isolate the rd1 cDNA.8 Similarly, Travis and co-workers used subtraction screening to obtain the rd2 (rds; retinal degeneration slow) cDNA.9 The rd1 gene is now known to be the β-subunit of cGMP (cyclic guanosine monophosphate) phosphodiesterase,10 and rd2 is the peripherin/rds gene.11
Technical advances have made possible the analysis of large cDNA libraries. DNA sequencing technology has become rapid enough that large numbers of clones can be sequenced. The function of many of these cDNA clones remains unknown, but DNA and protein databases are large enough so that the function of many clones can be identified by comparing their sequences to those already in databases. Adams and colleagues, who were among the first to adopt this strategy, employed brain tissue cDNA libraries and accumulated more than 2500 unique expressed sequence tags (ESTs).12,13
Many genes and cDNA copies of mRNAs expressed in the retina and other eye tissues have now been cloned and sequenced. Groups such as the IMAGE consortium14 have sequenced large numbers of cDNA clones from a variety of tissues including retina, lens, cornea, optic nerve, and trabecular meshwork. These sequences have been entered into the National Center for Biotechnology Information (NCBI) database and are accessible through Unigene; the clones from which these sequences are derived are available through Open Biosystems (Huntsville, AL). In addition, some of these ocular expressed genes are stored as subsets of the complete genome in a separate database archived and maintained at a site called NEIBank.15,16,17,18,19 A consequence for ophthalmology is that large numbers of new eye-expressed genes have been discovered. Each of these genes is a candidate for a cause of inherited eye diseases, with the potential for highly accurate diagnoses. As these lesions are characterized and as the functions of these genes are discovered, the expectation is that potential therapies will be developed.
Genomic libraries are constructed in much the same way as cDNA libraries, except that the insert DNA originates from genomic DNA. Genomic DNA is isolated from any tissue from the body, either germline or somatic tissues. Because this DNA should be large, special care should be taken not to shear it during its isolation. To produce fragments approximately 20,000 bases in size, usually the DNA is partially digested with a frequent cutting restriction enzyme, such as Sau3A, a “four-base cutter” that recognizes GATC and digests or “cuts” before the first G. A vector, frequently based on bacteriophage lambda, is digested with a compatible restriction enzyme, in this case, BamHI, which recognizes GGATCC, cutting between the first and second G. The sticky ends of the genomic DNA and vector match and anneal when mixed together. The insert genomic DNA fragment is ligated into the vector, and the recombinant DNA is packaged into bacteriophage lambda with extracts containing phage components that can reassemble themselves very efficiently into an infectious particle. The particle infects E. coli, grows to produce more infectious phage particles and lyses the host cell, and the daughter phage continue the cycle. A phage particle is plated onto a lawn of E. coli growing uniformly over the surface of a Petri dish, and after a few hours a clear dot (a phage plaque) appears as a hole in the otherwise turbid lawn of bacterial growth. Each plaque (an aggregate of approximately 106 phage particles), which is generated from a single infection by one bacteriophage, is distinct and separate and represents a single recombinant genomic clone. Because there are approximately 3 × 109 base pairs (bp) per human haploid genome, millions of recombinant phages are required to achieve a complete representation of the genome. For some applications, clones containing larger inserts are needed. Cosmid vectors (40 kb inserts) and BACs and YACs (150-kb to 1-mb inserts), which are discussed later, fulfill this need.
Usually a cDNA clone is obtained before a genomic clone is sought. Thus, the cDNA clone is the most frequently used probe to screen a genomic library.
Positional cloning is the next most prevalent strategy used to find a specific genomic clone. Linkage analysis (described later) provides the location, usually to within approximately 1 to 5 million bases of the gene. To move closer to the gene, several techniques are applied. This approach, formerly called “reverse genetics” and now known as “positional cloning,” simply refers to a strategy using the known chromosomal position of the disease locus to isolate the corresponding gene. This is discussed in detail below.
MICROARRAYS
Northern blot and reverse transcriptase polymerase chain reaction (RT-PCR) assays are powerful tools in the analysis of gene expression levels. These techniques are straightforward, fast, and easy, provided that only a handful of genes need to be analyzed at one time. But what if an expression level analysis of many or even all genes needs to be conducted? A technique has been developed to analyze the abundance of thousands of mRNAs simultaneously. The principle of this technique, called microarray or GeneChip analysis, relies on the specificity of annealing a single mRNA type to a pure DNA sequence. Thousands of different DNA sequences are spotted individually onto a two-dimensional surface where each clone is bound irreversibly. This can be accomplished by spotting each cDNA clone onto a chemically modified glass microscope slide. Each spot contains picoliter volumes of a single pure cDNA. Another way to create the array of pure DNAs is to synthesize oligonucleotides directly onto the glass surface, using photolithographic processes similar to those employed for creating computer chips. Two commercially available chips prepared in this fashion represent over 33,000 genes, approaching the entire repertoire of the human genome. A third variant is to create a macroarray by spotting much larger spots onto nitrocellulose filters.
Once the array of the thousands of different cDNAs is created, the second step is to probe the array with labeled mRNA from the tissue or sample of interest. Several alternatives are available to tag the mRNA. Usually the marker is a fluorescent tag, providing excellent signal sensitivity. A key point is that two or more different probes are prepared. One probe is derived from an experimental condition, and the second probe serves as a control condition, which can be mock, sham, or vehicle treated. Many other experimentally different conditions may be used.
The labeled probe is actually a concoction of thousands of different labeled mRNAs. Each mRNA differs in abundance from the next, and the abundance of each reflects its relative abundance in the sample. The probe is incubated on the surface of the chip in a conventional hybridization mix. Under these hybridization conditions, usually about 52° C and 6X SSC, each different labeled mRNA will hybridize specifically to the fixed amount of DNA on the correctly mated spot on the array. The amount of label bound to the spot reflects the abundance of the mRNA in the sample. Any unbound probe mRNA is removed by washing, much as in Northern or Southern blot analyses. The array is “read” in an instrument capable of detecting the specific label being used in the experiment. The levels of each mRNA are compared in the experimental sample versus those in an identically processed control sample. Thus, in one experiment, which usually takes only about a day, the levels of mRNA accumulation for many thousands and even all genes can be measured.
The utility of this approach is twofold: First, the analysis of genome expression is encyclopedic, as virtually all known genes can be analyzed; second, the work need not be hypothesis driven. Although many National Institutes of Health (NIH) initial review groups find this frightening, horrifying, and appalling, a fishing expedition sometimes can be useful. That is, the global capabilities of microarray analysis may unlock hidden processes that the scientist had not previously envisioned. Microarray results may radically change hypotheses about the mechanism(s) of drug action and disease etiology and may uncover previously unknown metabolic or mechanistic pathways.
Along these lines, Friend and colleagues20 employed microarray analyses to predict clinical outcomes among patients with breast cancer. They analyzed the gene expression patterns of thousands of mRNAs derived from biopsies of primary breast tumors from 117 women. They obtained a “signature” (a pattern of expression from 70 different cDNA clones) from the overall collection of about 25,000 genes. This signature was strongly predictive of a short length of time before distant metastases occurred among patients who were lymph-node negative at the time of diagnosis. The optimized signature contained genes regulating the cell cycle, invasion, metastasis, and angiogenesis. This classifier outperforms the currently used traditional clinical indicators that include histopathology grade, tumor size, angioinvasivenss, and estrogen receptor expression status. The signature is relatively large (70 genes) and remarkably does not include some markers that, by hypothesis-driven notions, one would expect to be included, such as HER/Neu, c-myc, ER-α, and so forth. One important element of this study is in the prediction of which patient will benefit from treatment; the gene expression signature is at least as effective as existing clinical guidelines. The signature is much more effective in finding patients for whom adjuvant chemotherapy would have no benefit. Thus, the signature is currently the best way to prevent unnecessary treatment, which can cause harm in itself. Another implication of the study is that a tumor’s decision to spread is cast early, while the tumor is very small. Last, the several genes that are highly overexpressed in the poor prognosis signature may be excellent targets for chemotherapy for breast cancer patients in the “poor prognosis” category.
Expectations are that in retinal and macular degenerations, even when the precise lesion remains unknown, similar microarray analyses may provide signatures of genes indicative of severe or rapid visual loss. The identified genes in the signature may provide analogous targets for drug therapy that might slow or reduce loss of visual function. Although it is clear that the retina should not be biopsied, the results from developing these signatures should be helpful. Also, in animal models of retinal degenerations, across many diseases, we may identify common metabolic pathways that are responsive to drug treatment.
There are a number of potential problems with microarrays:
The labeling technique may selectively favor one mRNA over another, thus distorting the mRNA expression pattern.
Low-abundance mRNAs may not be detected, either because the mRNA is not in the sample or because the minute amount of the label when bound to the spot is below the level of detection.
Microarray analysis requires large amounts of starting mRNA. In some schemes, to make large amounts of probe, the mRNA is amplified by an RT-PCR step. This may cause a misrepresentation of one mRNA for another because of different levels of efficiency in the RT-PCR steps.
An important cDNA may not be present on the microarray. Thus, accidentally, the gene and mRNA cannot be analyzed even though the experimenter thought the gene was present.
Artifacts in the cDNAs. Unwittingly, two cDNAs may be fused and spotted onto the array. Thus, what should be two different signals are combined into one.
Surface effects of the chips. Most chips are flat, two-dimensional solid surfaces with the clone or oligonucleotide bound directly to the surface. Because of steric hindrance at the surface of the chip, the mRNA may be unable to bind to the DNA, and this may result in artifacts. Long cDNAs, long oligonucleotides, or the use of spacers can reduce this type of artifact. Also, a three-dimensional surface can allow for a solution-based hybridization and avoid the steric effects.
Mishybridization. The wrong mRNA may adhere to an incorrect DNA. Increasing the stringency of chip washing conditions can reduce nonspecific binding. Also, the selection of probes that lack partial homologies can reduce misbinding. Mutating the DNA can help to identify improper hybridization. Stringent controls for the preparation of probes is the first line of defense against improper hybridization.
Dynamic range. Only a small amount of DNA is spotted onto an array, and it is possible that the mRNA is in very high concentration relative to the amount of DNA on the spot and saturates it. To circumvent this problem, it may be necessary to run the experiment with two different doses of mRNA, a high probe concentration and a low one.
mRNA splice variants. Many genes can be alternatively spliced, and different tissues may favor one splice variant over another. One way to work around this problem is to employ two different DNAs on the chip, one specific for one splice variant and a second specific for the other variant.
mRNA expression levels are useful, but often the critical information may be the level of enzymatic activity or the amounts of protein present. mRNA microarrays measure steady-state mRNA levels, not levels of protein, enzyme activity, or even the rate of transcription of RNA. Thus, it is critical to know exactly the question being asked in order to decide whether microarrays for measuring mRNA levels are the appropriate technique. The correlation between mRNA level and protein level is about 70%.21
Replication. Because of the expense of microarrays, and because they cannot be re-used, there is the temptation to cut corners on controls and numbers of replicates. The same scientific standards apply to microarray analyses as to any other procedure. Because of the exceedingly large numbers of genes being analyzed, random coincidence may explain why some clones exhibit changes in expression during treatment.
Last, there are potential problems with analysis techniques, data storage and documentation, and standardization among reporting formats so that results across platforms and labs can be compared. In the past, only a handful of genes and mRNAs were being studied at the same time, and the reader could easily compare results. Today, however, thousands of genes are being analyzed simultaneously, and the reader must rely on computers and databases to compare results across several publications.
Despite these problems, microarrays exhibit numerous advantages that outweigh the known difficulties. It is an approach well-designed to exploit information encoded in the human genome sequence and is useful for basic and applied needs.
PROTEOMICS AND CLONING
A proteome is defined as the complete set of proteins produced from the information encoded as a genome. A proteomics-based strategy, based on a combination of mass spectrometry and database searching, has become popular for identifying a protein and obtaining its corresponding cDNA clone. Once the identity of the protein is established, because of the completeness of the human and other genome databases it is often possible to retrieve a cDNA or genomic clone from the original library source. The cDNA also can be obtained by PCR analysis (described later)
The proteomics approach usually begins with finding a spot on a two-dimensional (2D) protein gel that is the unknown protein of interest or that contains a unique biologic activity. 2D gels separate proteins based on the isoelectric point (pI) and molecular weight. An alternative is to resolve a peak by multi-dimensional chromatographic analysis. Often the protein is considered interesting by virtue of being differentially expressed in a developmental or disease state. Once the protein is sufficiently pure and the correct protein spot is found, it is isolated and digested with trypsin. The resulting proteolytic fragments are subjected to analyses in mass spectrometers.
The first such analysis is usually matrix-assisted laser desorption ionization time-of-flight (MALDI TOF) mass spectrometry (MS) This analysis provides experimentally determined masses of each tryptic peptide of the protein to within 0.01% accuracy. The set of masses of the numerous tryptic fragments are a characteristic “actual fingerprint” of the protein and, in principle, can be used to identify it. A database of known and predicted proteins has been derived from the human genome sequence. The masses of each set of tryptic peptides from each known or predicted polypeptide have been calculated from this protein database. The predicted set of masses from each protein in the database provides a “predicted fingerprint” to which the experimentally determined “actual fingerprint” is compared. We then ask whether the experimentally obtained actual fingerprint from the gel spot matches any of the thousands of predicted fingerprints in the database. Theoretically, one, and only one, protein should match perfectly, and this match provides us with the identity of the protein. It also provides us with the sequences of the protein, mRNA, and gene.
There are some drawbacks and potential problems with this approach. First, a critical assumption is that the resolution of the 2D gel is sufficient to assure that only a single polypeptide is contained in the spot. While 2D gel technology can resolve about 2000 different proteins on a single gel, this may not be sufficient resolution if contaminating proteins have nearly the same mass and pI as the protein of interest. Second, the abundance of the protein under consideration may be very low, and it may be necessary to spend substantial time and effort to obtain an enriched protein preparation such that the single spot of interest has no contaminants in it. Third, it is unlikely that every tryptic peptide is extracted from the gel spot. Thus, not every theoretical peptide mass will have a match to a peptide in the experimental data set. Next, it is inevitable that proteins from skin and other sources will contaminate the experimental sample, creating extraneous peptide masses that can confound analyses. However, if we find many peptides matching within 0.01%, this agreement suggests that the unknown protein of interest has been identified. Scoring systems that assess the quality and extent of the matches help to judge the probability that the identified match is authentic, and web-based utilities such as Protein Prospector perform these calculations. If the z-score is above 1.65, then it is highly likely (odds > 95%) that the match is correct.
Finally, much additional work is required to prove the putative identity. Sequencing the unknown protein by Edman degradation or a sequencing based on b- and y-daughter ion series from MS–MS experiments22 is, at a minimum, a firm requirement to establish and validate identity of the protein. Sometimes proteins are post-translationally modified. Under these circumstances the identification of the modifications may be made by additional MS methods22.
Once the protein is identified, in many cases, a cDNA or genomic clone corresponding to the newly identified protein can be obtained merely by ordering the clone from one of several commercial repositories.* A caveat is that these clones may not be complete. However, within days or even hours of identifying the protein of interest, it is possible to obtain at least a partial cDNA or genomic clone.
BIOINFORMATICS AND DNA SEQUENCE ANALYSIS
To acquire the information that a gene contains, we need to determine the sequence of bases in it. The entire human genome has now been sequenced, providing a database of all the approximately 35,000 genes in the human body, a molecular anatomy of the human genome. The sequences of this genetic material are now held in several commercial and public databases.
While there are still some gaps and little of the normal variability of the genome has been assessed, the availability of this sequence has changed the way in which positional cloning is carried out. The genomes of several additional organisms are also near completion. These provide a vast warehouse of useful information that ultimately will be employed in (1) elucidating the specific functions of all the genes, (2) diagnosing disease, and (3) designing treatment of retinal and many other diseases.
Because new sequence entries are shared among databases, submission of an entry to one database should assure that other databases receive a copy of the entry. It is no longer practical to analyze or compare sequences manually. With the devlopment of the field of bioinformatics, DNA sequences are now stored and analyzed by software suited to the given task. Many program packages are available for use on mainframe and personal computers, and several programs are available on the Web for sequence analysis.
For example, an investigator might need to retrieve all sequences expressed in the photoreceptor. Appendix I shows a partial list of sequence entries in the NCBI-GenBank database (Version 0.2), which were retrieved from the database with the keyword “photoreceptor” using the computer program “STRINGSEARCH” in the GCG program package. Although many entries are detected, other entries related to phototransduction are not listed. Thus, a search must be carefully constructed to avoid missing relevant sequences. A typical sequence entry (in truncated form) is shown in Figure 5. Besides the DNA sequence, a certain amount of header information is supplied, making it easier to identify how the sequence was obtained and to find features within the sequences that may be biologically important. An example of information from NEIBank is shown in Appendix III. This partial printout shows abundant cDNAs found in the retina.
 Fig. 5. A typical GenBank database entry. The entry for the human opsin gene is shown, displaying the header information provided for the typical entry and below the DNA sequence of the opsin gene, truncated for the sake of brevity. |
DNA SEQUENCE DETERMINATION
Two major methods are used for DNA sequence determination, and both techniques rely on the ability of electrophoresis in polyacrylamide gels under denaturing conditions to resolve DNA molecules differing in length by only one nucleotide. The electrophoresis technique can resolve DNAs differing by one base up to 1200 nucleotides in length, but for practical considerations, usually lengths up to about 600 nucleotides are analyzed. Depending on throughput needs, the gel may be in the form of a thin slab of cross-linked polyacrylamide formed between two pieces of glass; alternatively, thin capillary tubes are filled with linear polyacrylamide, and these serve to fractionate a set of DNA molecules. The latter is more amenable to high throughput analyses but requires a substantial cost in instrumentation. The former is the original method of choice but requires more set-up and hands-on time.
The two DNA sequencing principles are the Maxam and Gilbert24 approach, exploiting partial chemical degradation of DNA molecules, and that of Sanger and co-workers,25 making use of enzymatic reactions that synthesize DNAs of various lengths. Both techniques produce a nested set of related DNA molecules. These sets of DNAs begin at a common end and are identical in sequence, except they differ in size by a one-nucleotide increment at the other end. The synthetic steps of the Sanger method are illustrated in Figure 6. A template DNA is copied by a DNA polymerase that must initiate synthesis from a short oligonucleotide primer (usually 15 to 20 bases) that hybridizes (anneals or forms base pairs) with part of the template DNA. The enzyme requires the four deoxynucleotides, appropriate buffers, and Mg++ ions for the synthetic reaction to take place. Sanger’s key idea was the incorporation of a mixture of dideoxynucleotides and deoxynucleotides into the growing chains of DNA. The absence of a 3′ hydroxyl group in the dideoxynucleotide prevents the chain from elongating any further, terminating its growth once a dideoxynucleotide is incorporated. This termination generates a DNA chain of a distinct length. By mixing appropriate ratios of a deoxycytosine triphosphate and its corresponding dideoxynucleotide, dideoxycytosine triphosphate, a spectrum of DNA chains is produced with all chains starting at one spot (determined by the location of the primer) but with some chains terminating at each cytosine in the sequence.
 Fig. 6. DNA sequence analysis by the chemical synthesis method of Sanger.25 All genetic information is encoded in the sequence of bases of DNA. The method developed by Sanger and colleagues is outlined here. A pure source of template DNA and a pure primer that anneals to the template are required. Synthetic reactions make copies of the template DNA. An amount of dideoxynucleotide is included in each of four separate reactions to terminate the newly synthesized DNA chains. In the figure, the terminators are represented by the Greek letters α, γ, ε, and τ, which represent dideoxyadenosine, dideoxycytosine, dideoxyguanosine, and dideoxythymine, respectively. In each reaction, the ratio of terminator to normal deoxynucleotide is tuned so that for each reaction adequate amounts of terminated chains from 1 to approximately 400 bases in size are synthesized. These chains also are radioactively labeled during this synthesis step. We resolve these chains by running a urea-polyacrylamide gel, which is capable of resolving DNA molecules differing in size by only one base. We load the gel with the four different reactions side by side. After electrophoresis for a few hours, we obtain an image of the radioactive bands by autoradiography. The pattern of bands looks like a ladder. The bands at the bottom are the shortest DNA molecules, and we “read” the DNA sequence by starting at the bottom of the gel and stepping up one band at a time, shifting from one of the four lanes to the next, moving to the immediately higher band. The direction of synthesis is always 5′ to 3′, and the order from bottom to top of the gel is consequently also 5′ to 3′. |
Separately, three other syntheses are carried out for each of the other deoxynucleotide and dideoxynucleotide pairs. This produces a full set of DNA fragments, all starting at the same point but ending at each base position of the DNA fragment being sequenced. Because of the small amounts of material being synthesized, a chemical label, or tag, is incorporated into the DNA during synthesis. For small-scale projects, the usual tag is radioactivity, which allows the DNA to be detected by autoradiography. The isotope most commonly used for DNA sequencing is 35S; it emits a β-particle that interacts with the emulsion of x-ray film placed on top of the sequencing gel, with the resulting image of the gel shown on the autoradiogram. For larger-scale sequencing projects (and increasingly even small projects), automated systems using fluorescent-labeled nucleotides now allow rapid and highly efficient sequencing of large amounts of DNA; hundreds of automated fluorescence-based sequencers were used to sequence the human and mouse genomes.
The principle of chemical degradation sequence determination is partial degradation of DNA in four separate reactions, with each reaction specific for a different base. DNA is first labeled with a radioactive tag at one end and then subjected to the four degradation reactions. The reactions break the DNA at one specific base per strand. Because excess nonradioactive carrier DNA is included in the reactions, only partial degradation of the radioactive DNA occurs, ensuring that chain cleavage is random along the length of the DNA chain. The degraded chains form nested sets of molecules differing in length by only one base, and the products can be analyzed by acrylamide gel electrophoresis. Otherwise, the analysis of the products of the Sanger and the Gilbert method is very similar.
Sequences larger than the typical 600-base reading are obtained by assembling overlapping sequence readings. The overlapping readings can be obtained by using a different primer, subcloning different DNA fragments, or using another trick, such as transposing a primer annealing site into other parts of the DNA insert. The redundancy inherent in overlapping sequence readings contributes to the accuracy of the completed sequence. To further heighten the quality and reliability of the sequence information, the sequence of each strand of the DNA is determined. Each strand complements the other; that is, an A on the first strand always forms a base pair with a T on the complementary strand, and C pairs with G on the other strand. Mistakes in interpreting gel readings or other ambiguities (sequence artifacts) from one strand usually do not occur at the corresponding position of the other strand. On occasion, the accurate determination of certain sequences remains troublesome, for example, regions very high in G + C content, but several methods have been devised to read these areas. These include the use of different polymerases, higher temperatures, and nucleotide analogs to aid in the sequencing elongation reaction. Also, higher temperatures during electrophoresis and inclusion of more potent denaturation agents in the acrylamide gels can help to prevent unusual structures in the DNA that may cause incorrect mobility in the gel or capillary. Last, a switch from the Sanger to the chemical degradation method may aid in determining the sequence. Sequences should be greater than 99.9% accurate.
ALTERNATIVE SEQUENCING METHODS
Other methods26 for DNA sequencing have been suggested, and three of these are worthy of discussion here. Common attributes of the three include (1) the analysis of single molecules of DNA, obviating the need to amplify or clone DNA prior to sequence analysis, and (2) no requirement for chemical degradation or synthesis to obtain a nested set of DNA molecules.
In atomic force microscopy, one of these methods, because the four bases of DNA are shaped slightly differently, an atomic force microscope trace of a fragment of DNA theoretically can be used to decode the sequence.27 This technique currently resolves DNA of about 10 bp lengths,15 which is not sufficient for sequence analysis. Progress has been made to enable haplotyping of alleles.16
The second method employs a lipid bilayer containing a pore of hemolysin. DNA is driven through the pore by an electric field. Ideally, as each base passes through the pore there should be a slight fluctuation in current characteristic of the base that is passing at that instant. The sequence could be read by monitoring these small current changes with time.17 As yet, however, only short homopolymeric sequences can be differentiated.
The third method relies on the hybridization of very short probes to a single long DNA molecule. The probes currently are tetramers labeled with a fluorescent tag. On average, any given tetramer will hybridize to DNA every 256 nucleotides, but due to the vagaries of any sequence, the interval between each complementary binding site of the tetramer varies, just as the pattern of restriction sites generates different-sized DNA fragments. Unlike restriction site analysis, however, the present system simultaneously determines not only the length of each interval between tetramer binding sites but also the order of these intervals. Figure 7 shows an example. With 256 different tetramers, every nucleotide in any sequence would be covered by hybridization. In principle, large genomes could be sequenced by this hybridization approach.
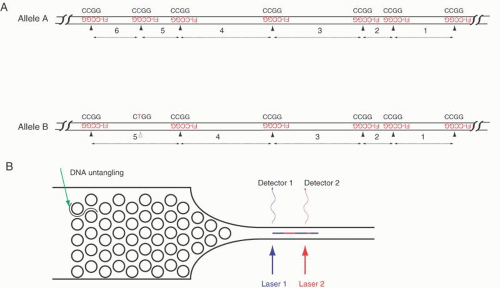 Fig. 7. Polymorphism detection in a single DNA molecule. Panel A. A fluorescently tagged tetramer (Fl-CCGG in red) is annealed to a genomic DNA fragment. Two different alleles are illustrated, with Allele A having one extra site and Allele B lacking one site. For the sake of clarity, only tetramer binding sites on the upper strand of DNA are illustrated. In comparing two alleles of the same fragment, sequence variations can be detected. Allele B has exactly the same fragment size and order as Allele A for the first 4 fragments, but fragment 5 is larger in Allele B, and Allele A contains two fragments (5 and 6) in place of Allele B’s single fragment 5. A critical point is that the former two from Allele A add up to the same size as Allele B’s fragment 5. Panel B. The hybridized probe-DNA complex is straightened in the funnel by catching on several pegs. The DNA passes by one or more fluorescence detectors. The DNA molecule moves across the detector at a constant rate of about 1 cm/sec. Because the DNA is straightened and in line with flow, the time elapsed from the detection of one fluorescent probe to the next reflects the number of bases between two adjacent tetramer binding sites. The order and sizes of the fragments can be recorded and compared with the predicted pattern of tetramers from the known human sequence. This identifies the location and length of the fragment. |
This method begins with hybridization of tagged tetramers to isolated DNA, followed by determination of the positions of the hybridization signals. By passing a single DNA molecule lengthwise through a detector, the positions of the hybridized tags along the DNA are recorded. The steps of the technique are, first, to prepare genomic DNA or DNAs from BAC or YAC clones by conventional means. Second, labeled tetramers of the same sequence are hybridized to a long DNA fragment, resulting in a set of tags along the DNA. Third, the tagged DNA molecule is untangled by passing it through a flat funnel-shaped microchamber containing a regular array of tiny posts. Each post is about 100 nm in diameter (the chamber is formed using the approach of nanotechnology similar to those used to manufacture silicon chips). The DNA collides with and is transiently caught on one of the many posts, stretching the DNA to either side of the post. As the DNA is pushed forward, it untangles a bit and falls onto another post. The series of collisions with several posts eventually untangles the DNA, rendering a linear molecule oriented lengthwise in a narrow channel at the far end of the funnel. The diameter of the channel is fine enough to keep the DNA correctly oriented, that is, straight and free from loops or crumples.
Fourth and last, positive pressure forces the DNA through the channel at a uniform flow rate, past a series of lasers, which excite the fluorescently tagged tetramers as each site on the DNA molecule passes by a detector. The time interval between two fluorescence events indicates the number of bases between two adjacent tetramer binding sites, and, in a long sequence, many intervals between tetramer binding sites are identified. In comparing the ordered set of intervals to the known human genome sequence, the particular DNA fragment is identified. An important byproduct of this analysis is that any missing or extra tetramer binding sites (besides the ends) suggest a sequence variation in much the same way as the appearance or absence of restriction enzyme site in RFLP analysis.
This method should allow rapid polymorphism typing, and it specifies which polymorphisms are inherited on the same chromatid. A second advantage of thistechnique is the rapid flow of DNA past the lasers and detectors. Currently, a DNA strand moves past the detector at a linear flow rate of 1 cm/sec (or about 30 million bases/sec). At this rate, the entire genome from one individual could be analyzed in 100 to 200 seconds. A third advantage is the tiny amount of DNA that is required, suggesting that cloning or DNA amplification may not be necessary. A fourth advantage is the possible development of multichannel instruments, because the techniques for creating funnels and microchannels appear to be amenable to parallel microfabrication.
Current disadvantages include the requirement for an entire collection of 256 probes to determine a full sequence. Although simultaneous hybridization with several tetramers can be conducted by employing several different tags that fluoresce at different wavelengths, it appears infeasible for a full set of 256 probes to be hybridized and analyzed simultaneously in a single run. It may be possible to reduce the size of the tags from 4-mers to 3-mers by using nucleotide analogs that melt at higher temperatures, concomitantly reducing the total number of probes from 256 to 64. Another problem is that DNA fragments that are too short may tumble in the microchannel; therefore, there is likely a minimum acceptable length of the DNA to be analyzed. From an efficiency standpoint, the DNA molecule should be as long as possible. However, it is easy to mechanically shear high molecular weight DNA, and it may be difficult to untangle DNAs larger than a million base pairs without breaking the DNA with the post method. Routine sample preparation, prior to introduction into an instrument, also can break DNA. Special precautions are necessary to prevent shear forces as DNA is isolated from cell nuclei. Last, it is not clear how a knotted DNA could be eliminated or excluded from analysis.
Despite the disadvantages of these new DNA sequencing approaches, they all exhibit desirable features, and in the long run they may vastly reduce the time and expense of current DNA sequencing techniques. They offer the promise of “personal sequencing” in which a single individual’s genome could be completely analyzed to test for a host of genetic diseases and hereditary risk factors or traits. Many areas of the genome are currently difficult to sequence because they cannot be cloned. Other sequences are difficult to sequence because present-day polymerases stall in attempting to pass through these DNA stretches. Other regions contain many repeats that are difficult to piece together by current sequence-overlap assembly methods. The three methods discussed here largely circumvent these common sequencing problems, and these new approaches should enhance the quality of the human and model genomes and their databases.
DNA RESEQUENCING
In some circumstances, both in basic research and clinical practice, one might not wish to sequence large parts of the genome, but might rather wish to sequence the same DNA multiple times. That is, one might need to screen a large number of patients for potential mutations in one or a few genes associated with a disease. It is possible to carry out screening using a variety of technologies based on the differing secondary structures caused by changing even a single base in a DNA sequence, such as single-strand conformation polymorphisms (SSCPs), denaturant gradient gel electrophoresis (DGGE), and DNA analysis using high-pressure liquid chromatography (HPLC) (these techniques are described in detail later in this chapter). However, these techniques have differing rates of detection of sequence changes and also require sequencing of the DNA once a variation is found.
In some cases, especially those in which very large numbers of samples are to be analyzed and frequent and variable sequence changes are anticipated, it is more efficient to proceed directly to sequencing each sample. Although this can be carried out using standard technology described previously, even high-throughput DNA sequencing is relatively expensive and somewhat inefficient compared to a true screening technology. However, the microarray approach described previously provides a potential answer to this difficulty.28 One can spot (or use photolithography to synthesize) overlapping oligonucleotides advancing by a single base to cover a whole gene (or several genes) on a DNA microarray. Each oligonucleotide is present in quadruplicate, with each possibility for the central base represented in a separate position. Then, if the microarray is hybridized with a copy of the target gene labeled with fluorescent nucleotides and washed under stringent conditions, hybridization to the exact sequence match will be highly favored over that of oligonucleotides containing a mismatch. By comparing hybridization to each possible nucleotide at each position along the DNA to be analyzed, the entire DNA sequence can be obtained with high accuracy in one hybridization. Current technologies allow sequencing of up to 60 kb on a single chip, making this a very competitive technology for large projects. A variation of this technique uses primer extension with fluorescently labeled nucleotides to provide corresponding sequence information.29
POLYMERASE CHAIN REACTION
The PCR is a favorite tool in molecular biology. The PCR technique makes practical the analysis of trace amounts of patient material, and reliable results are available in a short time (1 to 2 days). In some circumstances reliable data can be obtained in less than an hour. This technique has extended our analytic capabilities of DNA more than any other.30,31 PCR analysis amplifies a discrete sequence without the need to clone the DNA fragment. PCR techniques can amplify a sequence by a millionfold, allowing analysis of just a few molecules of DNA. When combined with other techniques, it allows sequence analysis, cloning, and almost any other enzymatic manipulation of the amplified sequence. It has made possible the concept of sequence tagged sites (STSs) (described later). DNA specimens suitable for use in PCR assays can be obtained from almost any tissue: blood, parts of histologic sections, buccal mucosa, hair follicles, anything with a nucleus, or in the case of a mitochondrially encoded gene, any tissue remnant with mitochondria. Ancient DNA samples, up to thousands of years in age, have been used as well. With adequate precautions to avoid contamination, the analysis of DNA from a single cell can be accomplished.
The basic technique for the PCR assay is shown in Figure 8. Amplification of sequences with PCR analysis is a simple concept. Two specific oligonucleotides, the “primers,” base-pair to opposite strands of a DNA sequence called the “template.” The primers are about 20 bases in length, are usually less than 10 kb apart, anneal to opposite strands of the template, and are oriented 5′ to 3′ pointed inward. The primers allow a DNA polymerase to initiate synthesis of new DNA that is a complementary copy of the original DNA strand. The primers are mixed with the template DNA (usually total genomic DNA containing the target sequence) in an appropriate reaction buffer with a heat-stable DNA polymerase isolated from thermophilic bacteria. The template DNA is denatured by heating the sample to 94°C, and the temperature is dropped to an “annealing temperature,” an experimentally optimized temperature at which the primers begin to anneal to the template DNA. This temperature is usually near the theoretical Tm, which can be calculated based on the DNA sequence. The Tm is the temperature at which 50% of a primer is base-paired to its complement and 50% remains single-stranded in solution. Because the oligonucleotides are present in vast excess, they successfully compete with one template strand for the target site on the opposite strand. The oligonucleotide primers are extended or elongated, usually at 72°C, oriented 5′ to 3′, copying the sequence of the target DNA. The reaction is conducted for about 30 sec at each of the three temperatures. The cycle (consisting of the three essential steps—denaturing, annealing, and elongating—in order) is repeated 20 or more times. Theoretically, each cycle doubles the number of target sequences, because newly synthesized DNA fragments can then act as templates for the primers. The PCR is exponential until reagents for DNA synthesis in the reaction run out.
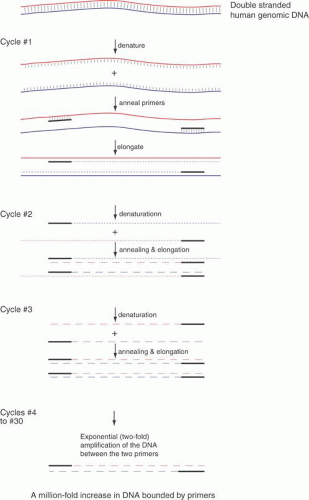 Fig. 8. The polymerase chain reaction (PCR). Amplification of a small amount of DNA from any of several different tissue sources by PCR assay has proved to be an invaluable aid to modern molecular biology and genetics. The steps in the PCR process are shown here. The reactions use several different temperatures to allow first denaturation, melting, of DNA strands from their normal double-stranded state into single strands. The second step is to anneal synthetic primers that bind to the melted DNA. The third step is to elongate and copy the template strands into new DNA in an elongation step. We cycle among the three steps at three different temperatures. Each temperature favors a different property: denaturation at 94°C, annealing at about 55°C, and elongation at 72°C. Usually 25 to 40 cycles are sufficient to synthesize enough DNA for easy visualization of the product by staining with ethidium bromide. In cycles beyond the third one, the PCR accumulates the desired product in exponential fashion. The first three cycles accumulate some newly synthesized DNA strands that extend beyond the end of one or the other primer, but these are of little consequence at the end of the amplification. |
Because each primer provides the same beginning, all the amplified products have uniform ends. However, Taq polymerase adds an extra unpaired A. This property is used to subclone fragments into a vector with a complementary protruding T.19
The fundamental limitation of PCR analysis is the requirement for sequence information on either side of the DNA sequence of interest. In certain circumstances only sequence information from one end of the region of interest is needed, but for optimal results and in typical PCR conditions, it is best to have a sequence from both ends of the region to be amplified. Because of the specificity and fidelity of PCR analysis, this amplification reaction can replace far more tedious recombinant DNA techniques, including cloning, library screening, and Southern blot techniques. The products of the PCR can be used to analyze and define gene defects and to define and use polymorphic DNA sequence variations. Analysis of the samples after PCR amplification is easy and uncomplicated. The PCR product can be sequenced directly. It can be analyzed by any of several electrophoretic techniques designed to exploit variations in the mobility of DNAs (discussed later). Finally, the amplified sequence can be subcloned and used in any way that any other cloned DNA fragment might be used.
Many creative embellishments have been coupled to the PCR technique. To quantify small amounts of a specific RNA found in tiny amounts of tissues or tumors, the PCR can be coupled to the reverse transcription of RNA. For example, a hundred cells laser-captured from the photoreceptor layer of the retina can serve as a source of RNA. RNA isolated from the cells is copied into cDNA by reverse transcriptase. The cDNA can then be amplified by PCR analysis. If fixed quantities of standards, either RNA or DNA, are similarly treated, the amount of an mRNA can be calculated. These measurements can be made on the fly. An example is shown in Figure 9.
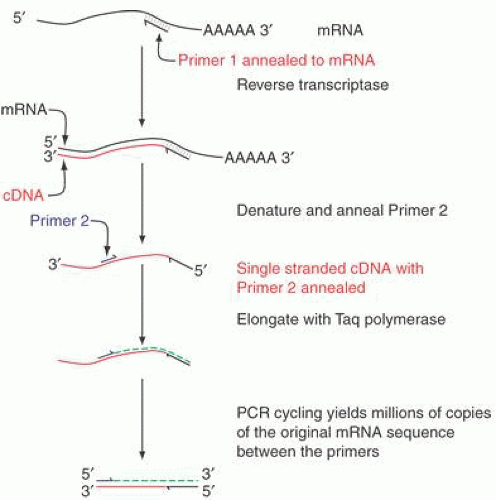 Fig. 9. Reverse Transcriptase coupled to PCR. One of the most important uses of the PCR is to measure the amounts of a specific mRNA. Changes in the accumulated levels of a specific mRNA can be highly diagnostic of disease processes, for example in the prognosis of breast cancer.20 To accurately and precisely measure the level of a specific mRNA, total RNA from a tissue is copied into cDNA with reverse transcriptase and a specific primer (Primer 1 in the illustration). A second specific primer (Primer 2) is used to initiate elongation of the cDNA with Taq polymerase or a related temperature-stable polymerase. A double-stranded cDNA results from these two steps, and the same primer pair is used to continue the PCR. Provided that the reaction is not cycled so many times that the substrates are depleted, the amount of PCR products is proportional to the starting concentration of the mRNA of interest. By carrying out simultaneous reactions with standard amounts of the specific mRNA, a calibration curve can be generated to calculate the absolute amounts of starting mRNA. This allows precise measurements of the changes in mRNAs in response to drug treatments, experimental manipulations, or disease courses.183 |
Single-base sequence variations can be analyzed by PCR assay. Suitable oligonucleotides flanking a base change are used to prime the PCR, amplifying the included region by as much as a millionfold. Within limits of approximately 100 bp to several kb, the size of the amplified fragment can be adjusted for convenience in the analysis, especially if multiple loci are to be analyzed simultaneously. Once the target sequence is amplified, it can be analyzed in several ways. Most directly, the PCR sample can be digested with an appropriate restriction enzyme for the base change, subjected to gel electrophoresis, and visualized by staining with ethidium bromide or other dye that fluoresces only when bound to double-stranded DNA. This provides an efficient and reliable analysis without requiring radioactive label. This technique is shown in Figure 10. Another approach is to employ one primer that contains a perfect match to one allele but that bears a single base mismatch at the 3′ end with the other allele. This primer provides a specific amplification only when the perfectly matched allele is present. By analyzing PCR product accumulation after each cycle, typing of an individual can be carried out. The patient’s status as a double homozygote for one allele, a homozygote for the other allele, or a heterozygote can be accomplished (Fig. 11).
 Fig. 10. A DNA polymorphism detected by a restriction enzyme. Individual A is a homozygote for one allele; individual B is a homozygote for the other allele. Individual A has a central Msp 1 site, CCGG, whereas individual B has lost the site due to a single base change of C to T, changing the recognized CCGG sequence to CTGG. On electrophoresis individual A has bands 1 and 2, while individual B has a single band, band 3, the size of which is the sum of the sizes of bands 1 and 2. Offspring of A and B are heterozygotes and have one of each of the alleles and all three bands. On Southern blot analysis with a probe indicated by the black bar, only bands 1 and 3 are detected. Band 2 does not contain sequences to which the probe can hybridize. Thus, on autoradiography (radiolabeled band) we will detect only band 1 in individual A, only band 3 in individual B, and bands 1 and 3 in individual C. |
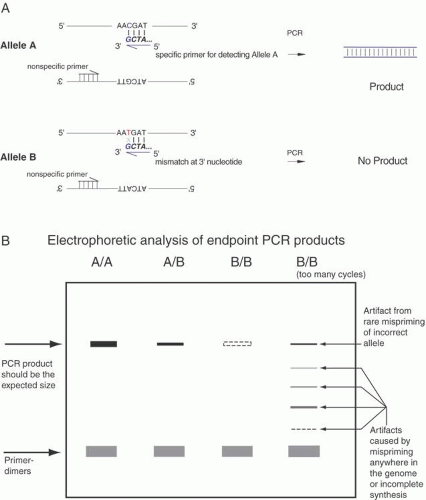 Fig. 11. Allele-specific PCR by endpoint analysis. Panel A. Allele A and Allele B can be distinguished by PCR assay provided that an allele-specific primer (with a perfect match to one allele and a single base mismatch at the other allele) is known. In the illustrated case, a 3′ terminal G matches perfectly to a C in the upper strand of Allele A, and the 3′ G mismatches with a T on the upper strand of Allele B. Under optimized conditions for the PCR, the reaction can be tuned to produce large amounts of PCR product from allele A with little or none from Allele B. Panel B. Electrophoretic analysis of PCR products from individuals who are homozygous for Allele A (A/A), heterozygous for Alleles A and B (A/B), and homozygous for Allele B (B/B). A heavy PCR band is expected for any A/A individuals, about half as much product is expected for heterozygous (A/B) individuals, and no product is expected from homozygous (B/B) individuals. The electrophoretic gel analysis allows the measurement of the size of the PCR product, and it should be the expected size. The product can be restriction digested to test whether the product has the anticipated restriction enzyme sites, and the product can be sequenced to further validate the its. Several possible artifacts of the PCR process are illustrated in Panel B. Primer-dimers can occur if the primers can bind to each other. Because of the high concentrations of the primers in the PCR, there is a propensity for the primers to bind to each other even when just few bases near the 3′ ends of the primers match each other. The primer dimers are detected near the bottom of the gel corresponding to the sum of the lengths of the primers, usually about 40 base pairs. Increasing the annealing temperature and decreasing the concentration of the primers may reduce the amount of primer-dimers. The far right lane [labeled B/B (too many cycles)] illustrates several other potential PCR artifacts. These are usually most noticeable when the PCR is subjected to too many cycles. In most cases, with 10 ng genomic DNA, a PCR is complete by 25 to 30 cycles. In certain circumstances it may be necessary to run the reaction for more cycles, and under those conditions a series of artifacts may be detected. One artifact is that after too many cycles, reagents and substrates will become rate limiting and products may not be completely copied. These partial products can be detected at many intermediate sizes, from very small to nearly full size. Also, after being subjected to repeated heating, contaminants in the reagents may reduce the specificity of the annealing, and mispriming can occur. This may lead to the formation of a full-ength PCR product, even in the presence of only Allele B DNA. Thus, the PCR must be optimized for the number of cycles to maximize the yield of the A/A product, minimize the amount of any B/B product, minimize primer-dimers, and minimize any incomplete products. The optimization process usually titrates the number of PCR cycles, annealing temperature, concentrations of primers, Mg++ ions, buffers, and type and amount of polymerase. Usually, several candidate primer pairs are tested. |
One limitation of the PCR technique is that the polymerase used sometimes misincorporates the wrong nucleotide. Thus, care must be taken when single PCR products are subcloned and analyzed by DNA sequencing or restriction analyses. At least three clones must be analyzed to ensure that the correct sequence has been obtained. When the entire PCR product is analyzed directly, this low frequency of errors is of little import. Several different polymerases are now available, including Pfu, Vent, and Hot Tub polymerases, which have lower error rates than Taq polymerase.
POINT MUTATION DETECTION
Point mutations are variations at one base. These can be detected if they fall in restriction enzyme recognition sites as described in the section on RFLPs or by several other techniques, also described in this section. Each of these techniques has advantages and has played a role in developing polymorphic markers in the past decade.
Orita and associates32 developed an approach for detection and identification of single-base changes called single-stranded conformation polymorphisms (SSCPs). Amplified DNAs are denatured by boiling and allowed to renature in dilute conditions that favor intramolecular interactions rather than allowing double-stranded complexes to form. Sequences that are identical except for a single-base variation may form similar, but not identical, secondary structures. Frequently, the free energy of the most stable structures may be very close, but the secondary structures may look entirely different. Inclusion of 10% glycerol, different detergents, or variation of the temperature at which a gel is run can markedly enhance the differences in mobility of two related DNA species. Although renaturation conditions must be optimized to maximize the differences in mobilities of two nearly identical sequences, once found, the technique is rapid and simple.
Heteroduplex analysis can be carried out directly after PCR amplification by the addition of one extra cycle of heat denaturation and slow cooling. In an individual who is a heterozygote for a point change, three types of annealed DNA duplexes will form. One pair of DNA strands will be homogeneous for allele 1 sequences (a homoduplex), another homoduplex pair will be homogeneous for allele 2, and the third pair will be a heteroduplex of one strand from allele 1 and the other strand from allele 2, as shown in Figure 12. The homogeneous strands of both alleles usually run with identical mobility on a polyacrylamide gel; however, the heteroduplex usually runs slightly slower. This decrease in mobility is due to the lack of the hydrogen bonding of bases at the point variation, creating a “bubble” in the middle of the double stranded DNA. Again, the conditions of electrophoresis can be optimized to maximize the mobility difference of the homogeneous DNAs compared to the heteroduplex structures.33,34
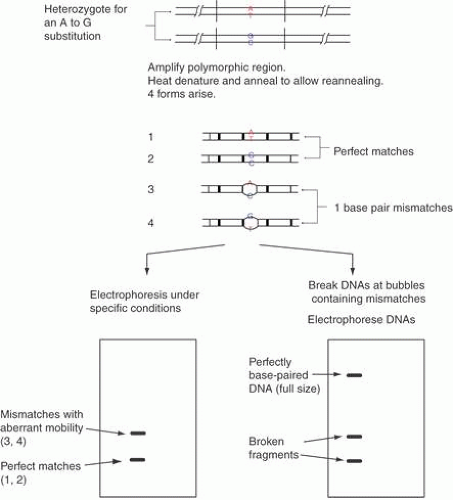 Fig. 12. Heteroduplex analysis: Detection of single-base variations, either mutations or polymorphisms. Suppose that an individual is a heterozygote for a single-base change, A to G. To detect the heterozygous state, we can use several different techniques, two of which are illustrated here. Assume that we amplify the region bounding the base change. By heating the resulting DNAs, we will obtain four different double-stranded DNAs; two of the four will be perfect matches, and the two others will be so-called heteroduplexes. The heteroduplexes contain single base–pair mismatches, and no hydrogen bonding occurs at the mismatch. Simple electrophoresis in some conditions allows us to resolve the perfectly base-paired DNAs from the heteroduplexes. This is illustrated on the left. The nonbase-paired heteroduplex bases are more susceptible to strand modification by reagents frequently used in Maxam and Gilbert sequencing. These weaken the DNA, and it is selectively broken at that spot by piperidine. The broken strands have very different mobilities on a urea acrylamide (sequencing) gel from the those of the homoduplexes. All possible substitutions can be detected with this method, although it requires extra steps. |
Newton and co-workers35 applied chemical degradation reactions to modify selectively the nonpaired bases of the heteroduplexes and then break the DNA strands at the modified bases.36 The heteroduplexes are cut into two or more smaller fragments of DNA, distinguishing heterozygotes from the homozygotes, as shown in Figure 13. A similar technique consists of hybridizing RNA complementary to one allele with sample DNA.37 The RNA-DNA hybrid is then treated with RNase A. If even a single base mismatch exists, the RNA will be cut, and the sample can be analyzed by gel electrophoresis. This is similar in principle to the analysis of DNA-DNA hybrids by modified Maxam-Gilbert sequencing reactions, in which strand breaks occur preferentially at mismatched bases.36
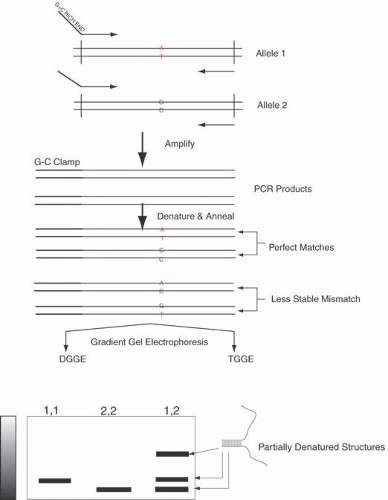 Fig. 13. Denaturing gradient gel electrophoresis (DGGE) and thermal gradient gel electrophoresis (TGGE). Another approach to analysis and detection of single-base changes is the exploitation of the difference in melting behavior resulting from a single base change. PCR-amplified products are used as before except that one primer contains an extra sequence at the 5′ end, which is G + C rich. This sequence when amplified is called a G-C clamp, and it melts at very high temperature in comparison to the typical sequence adjacent to it. In DGGE, the typical sequences will melt at varying denaturant concentrations along the solvent gradient. When they melt, the structure becomes Y-shaped because of the very stable G-C clamp. The resulting Y-shaped structure migrates extremely slowly in the gel. The gel contains a linear gradient of a chemical denaturant, urea, more concentrated at the bottom than the top. Thus, a double-stranded DNA migrates at its normal rate in a gel until it encounters a urea concentration high enough to partially denature the double-stranded DNA into the Y-shaped structure, and then it appears to stop. Different DNA sequences will migrate to different points along the urea gradient before converting to the Y-shaped structure. If two sequences that differ only by one base are run side by side, it is difficult to predict which will stop migrating sooner, but we might expect that one with an A: T base-pair would melt before an identical sequence excepting for a G:C base pair. However, heteroduplexes (double-stranded sequences containing one or more mismatches) with even a single mismatch of an A with a C or a G with a T would be expected to form Y-shaped structures at considerably lower concentrations of denaturant than strands that are perfectly matched and that bear the same flanking sequences. Similarly, in TGGE there is a temperature gradient within the gel increasing from cathode to anode; that is, the gel is hotter at the bottom than the top. The temperature gradient replaces the chemical denaturant gradient required in DGGE. The heteroduplexes melt at a lower temperature than the homoduplexes, at about 1.5°C per 1% difference; thus, a single-base change results in a significant change in the denaturation point. Following PCR assay, a heterozygote (shown here as an A to G substitution, in otherwise identical sequences) sample will contain four different pairs of sequences: two homoduplexes (1:1 and 2:2), and two heteroduplexes (1:2 and 2: 1). Homozygous individuals (1,1 and 2,2) are shown for comparison. Their predicated mobilities on DGGE and TGGE are indicated. |
Techniques dependent on melting hybrids or snapback regions of a DNA strand, such as denaturing gradient gel electrophoresis, have been useful.38 Denaturing gradient gel electrophoresis (DGGE) and thermal gradient gel electrophoresis (TGGE) are similar techniques designed to identify the differences of the heteroduplexes and the homoduplexes. Denaturing gradient gel electrophoresis allows reliable identification of single-base changes in a DNA fragment up to 1 kb in size. These techniques are illustrated in Figure 13. DGGE and TGGE are based on the principle that the heteroduplex, because of its one mispaired set of bases, is less stable than a homoduplex. By performing electrophoresis of these DNAs in an increasing gradient of either temperature (TGGE) or concentration of denaturant (DGGE), the heteroduplex will partially melt before the homoduplexes. Together with the property that Y-shaped and partially denatured DNAs are much less mobile than double-stranded DNA, this makes identification of heterozygotes straightforward. The stem of the Y-structure is engineered by a G-C clamp that provides a very stabile double-stranded region. This region is added by incorporation of a G + C– rich sequence at the end of one of the oligonucleotide primers. The heteroduplexes, becoming Y-shaped earlier during electrophoresis, appear as slower moving bands than the homoduplex samples that convert to a Y shape much later during the run.
Denaturing high-pressure liquid chromatography (DHPLC) has become a standard method for the analysis of point or short mutations in known sequences. Frequently these are polymorphic markers, but unknown mutations can be identified using the same approach. In DHPLC, PCR-amplified DNAs containing the polymorphism are allowed to bind to bind to a resin by ion-pairing. In an elevating temperature gradient, homodimers elute from the resin at certain well-defined temperatures and heterodimers elute at slightly different temperatures. These elution profiles are highly reproducible. Websites are devoted to the prediction of the best primers to use for optimum separation of the homo- and heterodimer complexes (see for example, http://www.insertion.stanford.edu/melt.html).
None of the previous methods requires à priori knowledge of the precise base change. If this information is available, other methods become very useful as well. Many polymorphic base changes do not occur in the recognition sequence of any restriction enzyme. These can still be analyzed by PCR assay with the use of allele-specific oligonucleotide (ASO) hybridization. In particular, differential hybridization can be used with mutant, wild-type, or allele-specific probes. Oligonucleotides of 17 to 19 bp in length are designed to be complementary to each allele at the variable site. These oligonucleotides usually vary at a single base near the middle of the fragment. PCR assay is performed on the sample DNA as above with the usual priming oligonucleotides, and the sample is subjected to gel electrophoresis and Southern transfer or to slot blot analysis. Hybridization is performed under permissive conditions, followed by a stringent wash step, which dissociates any mismatched complexes, allowing a positive signal only in the presence of the allele homologous to the labeled ASO. Temperatures for washing blots can be adjusted so that only hybridization of the perfectly matching oligonucleotide probe occurs. The difference in washing temperature can be determined empirically to make the differentiation and in practice is about 4 °C. With this technique, any single-base change in the genome can be analyzed efficiently and dependably.
Another means to detect variations is the use of an oligonucleotide with the 3′ terminal base as the mutation or sequence variant. This can be used to differentially amplify only one allele in the PCR. This is known as allele-specific PCR (ASPCR)39 or allele-specific amplification (ASA).40 Competitive oligonucleotide priming (COP) is closely related.41 Figure 14 shows an important variant of this approach that can provide more quantitative information on allele frequencies in pooled samples. This approach is called allele-specific real time PCR (ASRTPCR).
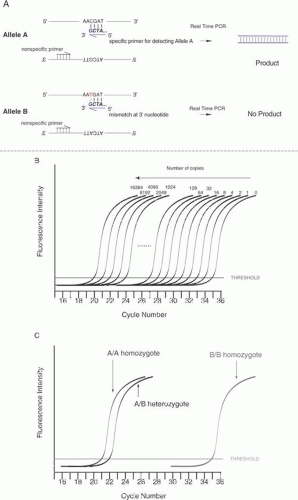 Fig. 14. Allele-specific real-time PCR. Panel A. A single nucleotide mutation can be detected by allele-specific PCR, if the single nucleotide change is known in advance. One of the two primers is chosen so that its 3′ nucleotide contains the variant single nucleotide unique to one allele. In this illustration, a single nucleotide change from C to T is the only difference between Allele A and Allele B. The second PCR primer matches sequence at both alleles nearby (usually no more than 150 nucleotides away) and is identified on the figure as the “nonspecific primer.” Because of the one-base mismatch between the Allele A–specific primer and the Allele B sequence, Taq polymerase will not synthesize a new DNA copy. In the Allele A DNA sample, the polymerase efficiently synthesizes a new DNA strand because the 3′ base matches this allele perfectly. |
In the primer extension assay, a primer is designed that perfectly matches the sequence immediately adjacent to, but not including, a single-nucleotide polymorphism (SNP). The genomic DNA from a sample is mixed with the primer and annealed. Polymerase and one of each ddNTP is added to each of four identical annealing reactions. The polymerase will add one dideoxynucleotide (ddNTP) to the primer depending on which allele is present in the sample. Heterozygote samples will allow primer extension in two of the four reactions. If the primer is radiolabeled, the primer and the elongated product can be resolved on a denaturing gel and the polymorphism detected by autoradiography. Recent improvements42 of the assay allow the mixing of the two relevant ddNTPs, and elongation with a polymerase is conducted in a single reaction. The two different homozygotes and heterozygotes can be resolved by DHPLC, despite the identical lengths of the two alleles.
Two of the chief methods (GeneChips and TaqMan) for SNP analyses are discussed in the next section.43
DNA POLYMORPHIC VARIATIONS
Polymorphisms are variations in the sequence of genomic DNA that have no obvious advantage or disadvantage for an individual and that occur at a frequency of more than 1% in a population. The analysis of variations in DNA sequences from one individual to the next has been particularly useful in ophthalmic genetics. Thousands of polymorphic variations have now been detected. In most cases, sequence changes are selectively neutral; they offer neither reproductive benefit nor detriment to the individual who carries the rare variation in comparison to the frequent allele. Usually a sequence change that results in a new characteristic or trait, for example, a change in function of an enzyme, is called a mutation. Sometimes mutations can be highly detrimental.44 The techniques described in this section allow one to examine individual variations in genomic DNA sequence and use these variations as markers in linkage analysis. Variations are now being cataloged in the NCBI SNP database (http://www-ncbi-nlm-nih-gov.easyaccess1.lib.cuhk.edu.hk/SNP/index.html). These variations fall into several classes and include microsatellites, restriction fragment length polymorphisms, variable number tandem repeats, point substitutions, and deletions, all of which are defined and discussed in this chapter. Detrimental hereditary sequence changes result in genetic diseases. Several thousand genetic diseases are known and have been cataloged by McKusick and colleagues. In Dr. McKusick’s on-line version of Mendelian Inheritance in Man (OMIM),† hundreds of inherited eye diseases are succinctly summarized, and clinical characteristics and disease etiologies are compared and contrasted.
SNPs
One class of sequence variation results from single base changes, which occur roughly each 500 bases and thus are the most common type of genetic variation. There are hundreds of thousands of these mutations in the human genome, and they are called single–nucleotide polymorphisms (SNPs). SNPs are stable and are inherited in a Mendelian co-dominant fashion. Unless these variations occur in an expressed sequence or in close-by regulatory sequences, they should have no impact on fitness, and selection does not occur for or against them.
SNPs provide one means of narrowing the linked region that must be searched to identify the causative gene. Because these small genetic distances are not amenable to linkage analysis, association studies are usually performed to identify likely candidate regions. As opposed to linkage, which identifies co-inheritance of marker alleles with a trait in families, association studies measure co-occurrence of a trait with particular marker alleles in populations. Thus, association studies are carried out in populations and depend on two things. First, the trait under study must have (1) occurred initially as a single “founder” mutation on a single chromosomal background and (2) been propagated through the population. Second, there must be a paucity of recombination events between the gene and marker, implying that only a very small distance separates them. If the disease to be studied originated from multiple mutations on many different chromosomal backgrounds, or if there has been significant recombination between the marker and the disease gene since the original mutation, there will not be significant levels of association in that population. It has been shown that the probability of recombination events in the human genome is not evenly distributed, but rather occurs at discrete areas, so that strong allelic association tends to occur between markers physically close to one another. The regions in which these groups of markers showing association occur are referred to as linkage disequilibrium blocks, or LD blocks.45 LD blocks tend to vary in size from 10 to 300 kb, having an average size of about 20 to 30 kb. On average, the blocks are somewhat larger in Caucasians than in Africans, probably because of the greater age, and hence genetic, diversity, of the African population. Usually, within a block the majority of individuals will display only a few haplotypes.
Because SNPs tend to be used in groups, and because they tend to be useful in projects in which large numbers of individuals must be genotyped, development of techniques for genotyping SNPs have tended to concentrate on high throughput methods.43 One method involves microarray analysis similar to that described for analysis of gene expression and resequencing. In this case, oligonucleotides encoding the polymorphic sites with all possible bases are arrayed on the chip. Genomic DNA from an individual is then amplified by PCR assay using primer pairs flanking the SNPs to be analyzed and a fluorescent tag. When the chip is analyzed, the oligonucleotide sequence containing the allele or alleles found in the individual being tested will show the strongest fluorescence. As an example of this technology, 1,500 SNPs may be tested at once using the Affymetrix GeneChip HuSNP Mapping Assay. An alternative method for analyzing SNPs uses the PCR assay and the 5’–3′ exonuclease activity of Taq polymerase. A hybridization probe is tagged at either end with a fluorophore and quencher. In this arrangement, the quencher decreases fluorescence by Förster resonance energy transfer. But under special circumstances, the nuclease activity of Taq polymerase can cleave the fluorescent tag from the end of the oligonucleotide probe and the free fluorophore is no longer quenched, producing a large fluorescence signal readily measured in real time. The probe is designed to hybridize perfectly to one allele, but is mismatched against other alleles. Inclusion of the oligonucleotide probe during PCR amplification allows the oligonucleotide to hybridize to its perfectly matching strand of DNA, and when annealed, the 5′-nuclease of Taq polymerase liberates the fluorophore. The assay mix also contains the alternative (mismatched) probe lacking fluorophore, but this DNA binds less well to the template DNA and is displaced rather than digested by the Taq polymerase as it progresses past the polymorphic site. When monitored in real time, the allele specificity of the perfect hybridization is observed, with a clear differentiation of homozygotes having two alleles matching perfectly versus heterozygotes having one copy of the allele and those having no copies of the specific allele. This type of assay can be designed specifically for any polymorphic site, or pre-designed assays are available, for example the Applied Biosystems Assays-On-Demand genotyping system. Approximately 2,000 SNPs can be tested with a throughput of over 5,000 genotypes a day using the ABI PRISM 7900HT Sequence Detection System. Additional methods of analyzing SNPs include the ligation chain reaction as described in Kwok and colleagues.43
RFLPs
A restriction fragment length polymorphism (RFLP, Fig. 10) is simply the gain or loss of a restriction enzyme recognition site by a variation within the recognition sequence. When a single-base change occurs in the recognition sequence of a restriction endonuclease, it can be detected by digesting genomic DNA with that restriction endonuclease. If the base change has altered the recognition site, the enzyme will not cut at that site, and a new DNA fragment equal in size to the two fragments flanking the site will be created. It is straightforward to detect these changes by Southern blot analysis using an appropriate probe. In the history of genetics, RFLP analysis was the major breakthrough that allowed to the first important linkage studies to be carried out. Before that time, only a few polymorphisms were detectable, including blood group antigens, human leukocyte antigen (HLA) markers, and protein electrophoresis mobility variants. Although these are highly polymorphic, they provide limited coverage much of the human genome. Consequently, only a handful of linkage studies had been conducted before RFLP analysis, and there was little confidence in the approach as a general method for finding defective genes. RFLP analysis almost immediately suggested the contrary, that linkage analysis would become a major method to find a gene defect. RFLPs were first detected by Southern blot analysis (see Fig. 2). Because bounds are set by nonpolymorphic common restriction enzyme sites at the ends of a fragment, on Southern blot analysis it appears that the sizes of the fragments are changing among the polymorphic alleles. However, the variation in this type of polymorphism is only a change in one or two bases in the restriction enzyme recognition site. More commonly, RFLPS are now detected by first PCR-amplifying DNA bounding the RFLP site, restriction digestion, gel electrophoresis, and staining to detect whether the restriction site is present.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


