Chapter 26 Metastatic, secondary and lacrimal gland tumors
Neuroblastoma and Ewing’s sarcoma account for most childhood orbital metastatic disease.1 Wilms’ tumor, testicular embryonal sarcoma, ovarian sarcoma, and renal embryonal sarcoma occasionally metastasize to the orbit.2
It is important to differentiate between blood-borne deposits of a malignant tumor (metastatic disease) and extension of a tumor into the orbital tissues from an adjacent structure (secondary disease). Retinoblastoma and rhabdomyosarcoma are the most important sources of secondary orbital disease in children (see Chapters 24 and 42).
Metastatic disease
Neuroblastoma
Neuroblastoma is the most common extracranial solid tumor of childhood accounting for 9% of all childhood cancers and is the third leading cause of cancer-related death in children. Its incidence peaks in infancy with a median age at diagnosis of 17 months. It arises from postganglionic sympathetic neuroblasts. Most primary tumors involve the adrenal medulla but they can occur anywhere within the sympathetic nervous system in paraspinal ganglia, neck, or pelvis. This is the most common source of orbital metastasis in children, accounting for 41 of 46 cases of orbital metastatic disease reported by Albert et al;1 but it is a rare cause of orbital disease representing only 1.5% of 214 orbital tumors reported by Porterfield3 and 3% of 307 childhood orbital tumors quoted by Nicholson and Green.2
Genetics
Only 1–2% of cases have a family history; the underlying genetic mutation has been determined for most of these pedigrees, helping to understand sporadic neuroblastoma. It arises from the interaction of multiple common predisposing genomic variations. The genetic characteristics of each tumor have important prognostic significance; high levels of MYCN (N-myc) proto-oncogene amplification are found in approximately 20% of primary tumors and are associated with a worse outcome for each tumor stage.4–7
Hyperdiploidy of tumor cell DNA content confers an improved prognosis for infants under 1 year of age at diagnosis. Conversely, segmental chromosomal alterations are associated with more aggressive disease.8 Many genetic abnormalities have been identified in primary neuroblastoma tumors but their independent prognostic significance remains unclear. The most frequent somatically acquired copy number abnormality is allelic gain of distal chromosome 17q, identified in over 50% of primary tumors. An unbalanced gain is associated with more aggressive disease and decreased survival. Cellular genomic aberrations are a better prognostic predictor of the tumor’s biological behavior than clinical factors such as age and stage at diagnosis. This is important for treatment planning since it is not uncommon for these tumors to regress spontaneously (stage 4S), even if already disseminated to the liver, skin, and bone marrow. If identified early, these infants can be spared the harmful adverse effects of chemotherapy since their survival rate exceeds 95%.9 In contrast, children with high risk neuroblastoma are often resistant to multimodality treatment and have a 5-year survival rate of 40%.
Clinical presentation
Most cases occur by 3 years10 and 90% are diagnosed by age 5, the range being from birth to the late teens. The adrenals are the primary site in 51% of cases, but the tumor can arise in the cervical sympathetic chain, mediastinum, or pelvis.11 Primary orbital neuroblastoma occurs mostly in adults.12,13 Neuroblastoma is more common in patients with neurofibromatosis type 1 (NF1).
The diagnosis is often not made until the patient has widespread metastases;14 40% have metastases at presentation, a proportion which rises to 55% in patients over the age of 1 year. Surprisingly, about 10% of tumors and their metastases (stages 1 to 4s) undergo spontaneous regression, something which occurs 100 times more commonly than for any other cancer.15 This fact underlies the cautious treatment approach outlined below.
Ophthalmic and orbital features
The presence of neuroblastoma in the mediastinum or cervical sympathetic chain may first manifest with Horner’s syndrome. This was the underlying diagnosis in two of 10 children with Horner’s syndrome reviewed by Woodruff et al.16 Gibbs et al.17 described congenital Horner’s syndrome in an infant with non-cervical neuroblastoma, suggesting that the two conditions might indicate a widespread dysgenesis of the sympathetic nervous system. Tonic pupils have been reported as a paraneoplastic effect of adrenal neuroblastoma.18 Iris19 and choroidal20 metastases from abdominal neuroblastoma have been described. The presence of opsoclonus (see Chapter 90), a striking large amplitude erratic ocular flutter also known as “dancing eyes syndrome,” with or without ataxia and myoclonus, suggests occult localized neuroblastoma.14 The primary tumor in these cases is in the chest or abdomen and not the brain. It is usually associated with a good prognosis, possibly because only single copies of the N-myc oncogene are present within the tumor cells.21 Opsoclonus can also be present with multiple N-myc copies, signaling a poor outcome.22
Presentation
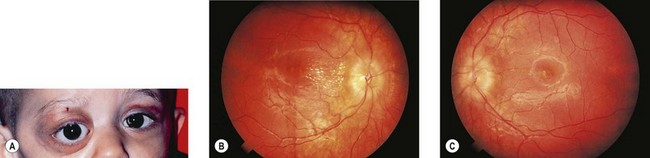
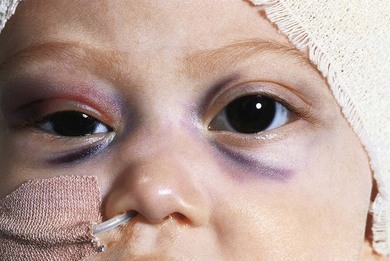
In 93% of the 46 cases reported by Albert et al.,1 the primary tumor had been diagnosed prior to presentation with orbital signs. Ninety percent of the 60 patients with orbital metastases reviewed by Musarella et al.14 had a primary abdominal tumor. Orbital metastases commonly present with sudden onset and rapid progression of proptosis (Fig. 26.1), unilateral or bilateral. Ecchymosis (Fig. 26.2) is present in 25% of cases.14,23,24 The lesion is usually in the superolateral orbit and zygoma but may occur anywhere within the orbit. Bony lesions give rise to swelling of overlying tissues so periorbital swelling and ptosis may be present. This presentation may be confused with orbital cellulitis or other rapidly progressive orbital tumors such as rhabdomyosarcoma, Ewing’s sarcoma, medulloblastoma, Wilms’ tumor, and acute lymphoblastic leukemia.25 A bleed into a pre-existing but clinically unsuspected venous lymphatic malformation (lymphangioma) may also present with sudden onset of proptosis and ecchymosis. The ecchymosis can lead to suspicion of child abuse and diagnostic delay.26
Treatment
The main prognostic (risk) factors are the age at diagnosis, stage of disease (Table 26.1), MYCN status, Shimada histology, and ploidy for infants. Survival rates for low risk groups are 90–100%, whilst those for high risk groups range from 20% to 60%.
Table 26.1 International neuroblastoma staging system (INSS)
| Stage | Description |
|---|---|
| 1 | Tumor confined to organ or origin |
| 2 | Tumor extends beyond organ of origin but not beyond midline |
| 2a | No lymph node involvement |
| 3 | Tumor extends beyond midline with or without bilateral lymph node involvement |
| 4 | Tumor disseminated to distant sites |
| 4s | Children younger than 1 year of age with dissemination to liver, skin, or bone marrow without bone involvement and a primary tumor that would otherwise be stage 1 or 2 |
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


