Metabolic Diseases and the Eye
Selwa Al-Hazzaa
Pinar Ozand
Elias I. Traboulsi
A landmark in the history of human biochemical genetics was set at the turn of the 20th century with Sir Archibald Garrod’s monographs on inborn errors of metabolism.
Metabolic and genetic disorders that affect the eye may cause significant visual disturbances and sometimes blindness. Furthermore, metabolic disorders may have characteristic ocular findings that assist in their diagnosis, making the ophthalmologist an invaluable member of the team that cares for these patients.
Metabolic disorders generally are inherited in an autosomal recessive fashion. There is reduced or absent function of one or more enzymes in a biochemical pathway that is critical to normal cellular function, growth, and development. Accurate biochemical diagnosis is essential for treatment, for genetic counseling, and for monitoring future pregnancies and children.
Clinically, enzyme deficiencies produce systemic and ophthalmic signs and symptoms by several mechanisms: accumulation of undegraded products, lack of production of an essential substrate, blockage of the normal conversion of one product to the other, or activation of alternate metabolic pathways that are deleterious to cellular integrity.
Inborn errors of metabolism can be divided into two broad categories4:
Category 1 consists of diseases that involve only one functional or anatomic system or affect only one organ. The presenting symptoms usually are uniform, and diagnosis is easy, even when the basic biochemical lesion gives rise to systemic consequences. Included in this category are bleeding disorders that result from coagulation factor defects or hemolytic anemia from defects of glycolysis.
Category 2 consists of diseases in which the basic biochemical lesion either affects a metabolic pathway common to a large number of cells or organs or is restricted to one organ but gives rise to humoral and systemic consequences. The presenting symptoms are diverse. This category includes most inborn errors of intermediary metabolism, diseases of intracellular trafficking, and lysosomal disorders.
Pathologic changes in the eye sometimes are characteristic of the underlying metabolic disease process. The detection of these ocular abnormalities depends on their prominence, the severity and ease of diagnosis of the systemic illness, and the familiarity of the ophthalmologist with their nature and significance. In many instances, the patient is referred with a diagnosed or suspected systemic disease, and a search for the known ocular complications of the illness is undertaken. In other instances, a metabolic disorder is suspected but no diagnosis is offered, making the ophthalmologic findings, if present, even more valuable in the diagnostic process.
Ophthalmologic findings such as corneal opacities, cataracts, cherry-red spot, and retinal degeneration may be the earliest signs of many metabolic disorders. Prompt and accurate diagnosis of the systemic disease aids in determining the prognosis and clinical expectations regarding career and life planning for the affected individual. It allows the early institution of treatment, when available, and the provision of genetic counseling on the risk of recurrence in siblings or in children. Measurement of enzyme levels or mutation analysis of DNA from fetal cells obtained through amniocentesis allows the prenatal diagnosis of many of these diseases.
Advances in molecular biology, biochemistry, and enzymology have allowed a better understanding of these diseases and their chemical defects. Currently, a biochemically based terminology is used instead of eponyms, and almost all disorders can be diagnosed by enzyme or gene analysis. Serum, leukocytes, or cultured skin fibroblasts can be assayed for enzyme activity if the molecular genetic defect is known, circumventing the need for biopsy of the liver, brain, or other tissues.
More than 300 human diseases that result from inborn errors of metabolism currently are recognized. Their true incidence may be underestimated because of failure of diagnosis. The detection of metabolic diseases relies only in part on screening programs and primarily depends on a high index of clinical suspicion and coordinated access to expert laboratory services.
Corneal opacities frequently occur in the mucopolysaccharidoses, mucolipidosis III and IV, α-mannosidosis, Fabry’s disease, multiple sulfatase deficiency (Austin disease), Farber disease, LCAT (lecithin: cholesterol acyltransferase) deficiency, cystinosis, tyrosinosis type II, and Tangier disease.
Cataracts are some of the more conspicuous ocular signs of metabolic disorders.5 At birth and in infancy, cataracts are prominent findings in Lowe syndrome. They can be the only presenting sign of sorbitol dehydrogenase deficiency. Cataracts commonly occur in peroxisomal biogenesis disorders and in Cockayne syndrome. They are the only signs of galactokinase deficiency. They also are present in various galactitol or sorbitol accumulation states of unknown origin. Cataracts can be associated with other typical systemic signs and symptoms in galactosemia, mannosidosis, sialidosis, respiratory chain defects, hypoglycemia due to galactosemia, the severe form of mevalonic aciduria, aspartylglucosaminuria, multiple sulfatase deficiency, and Fabry’s disease. In childhood, cataracts are present in one fourth of patients with untreated Wilson’s disease. They also are signs of hypoparathyroidism and pseudohypoparathyroidism. In adults, isolated cataracts can be a sign of Lowe syndrome or of the heterozygous state for inborn errors of galactose metabolism. They frequently are present in patients with cerebrotendinous xanthomatosis. Posterior subcapsular cataracts with onset in the second decade of life are nearly constant complications of gyrate atrophy of the choroid and retina.
A macular cherry-red spot is a characteristic finding in some lysosomal disorders. The ganglion cells filled with storage material in the macula are opaque and give rise to a white ring that encircles the red, ganglion cell-free fovea. The diseases associated with a cherry-red spot are GM1 gangliosidosis (Landing disease), GM2 gangliosidosis Type 1 (Tay-Sachs disease), GM2 gangliosidosis Type 2 (Sandhoff’s disease), Niemann-Pick Types A and B, sialidosis (cherry-red spot myoclonus), mucolipidosis I, metachromatic leukodystrophy, and galactosialidosis.
Retinal degeneration, with or without pigmentary retinopathy, occurs commonly in inherited metabolic disorders. Although its pathophysiology is not known, retinal dysfunction in metabolic disorders may be induced by toxic effects of certain metabolites, errors of synthetic pathways, or deficient energy metabolism. Table 1 lists the disorders associated with a pigmentary retinopathy.
 |
This chapter summarizes the systemic and ophthalmologic manifestations of metabolic disorders in which the enzyme deficiency results in visually significant or diagnostic ocular manifestations. Because the diseases of interest have etiologies and manifestations in complex overlapping, noncategorical biologic systems, the authors are faced with the classic dilemma of balancing the practicality of categorical thinking against the reality of biologic variation.
LYSOSOMAL STORAGE DISEASES
THE MUCOPOLYSACCHARIDOSES
The mucopolysaccharidoses (MPSs) are transmitted as autosomal recessive traits, with the exception of Hunter syndrome (MPS II), which is inherited in an X-linked recessive fashion.6,7 They are caused by deficiency of lysosomal enzymes needed for the degradation of mucopolysaccharides and glycos-aminoglycans.8,9 The catabolism of dermatan sulfate, heparan sulfate, keratan sulfate, and chondroitin sulfate may be blocked singly or in combination, depending on the enzymatic deficiency. The storage process eventually results in cell, tissue, and organ dysfunction. Glycosaminoglycan fragments are excreted in the urine.
A defect in 1 of 10 enzymes gives rise to each of the seven distinct clinical forms of MPS and their subtypes.Table 2 presents the classification, major systemic features, and ophthalmologic findings of the MPSs.
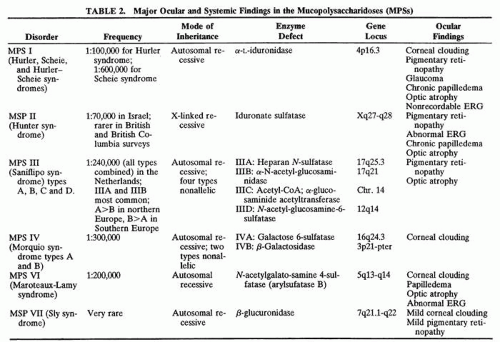 |
The MPSs share several clinical features that are variable in severity in individual diseases. These findings include skeletal abnormalities, coarse facial features, mental deficiency, cardiac disease, hepatosplenomegaly, ocular abnormalities, and deafness. A chronic and progressive course is typical. Ocular manifestations include progressive corneal clouding, retinal pigmentary degeneration, optic nerve head swelling, optic atrophy, and glaucoma.
Tissue-specific differences in the structure of mucopolysaccharides account for the variability in clinical manifestations. Excess dermatan and keratan sulfates appear in the cornea, whereas heparan sulfate accumulates in the retina and central nervous system (CNS). The accumulation of these substances is the result of the faulty catabolism of mucopolysaccharides in lysosomes due to defective lysosomal acid hydrolases. The large storage vacuoles in histiocytes, lymphocytes, or leukocytes show metachromasia.
In patients whose disease leads to the storage of heparan sulfate in the retina, a retinal pigmentary degeneration associated with night blindness develops. Early in the course of their disease, patients with MPS IH, IS, IH-S, IV and VI report moderately severe photophobia. Papilledema is a frequent finding, occurring in one third or more of patients with certain types of MPS, such as Hunter syndrome.10 Optic nerve head swelling has been attributed to the hydrocephalus that results from meningeal thickening with the storage material. Collins et al10 postulated that it could be caused by narrowing of the scleral canal at the optic nerve head, as a result of posterior scleral thickening with mucopolysaccharide accumulation. Acute and chronic glaucoma may be seen in MPS IH, IS, and IH-S.
Conjunctival biopsy, a simple and applicable procedure, is a reliable screening test for patients with suspected lysosomal storage diseases.11 The diagnosis of MPS is based primarily on the characteristic clinical findings and on the detection of mucopolysaccharides in the urine. For definite diagnosis and for further categorization of the types and subtype of MPSs, specific enzymatic assays or gene analysis should be performed.
HURLER SYNDROME (MPS I-H)
Infants with Hurler syndrome appear healthy at birth. Signs of progressive organ dysfunction and dysostosis multiplex appear in the first year of life. Hurler syndrome has been the prototype for the description of the MPS and is the most severe form, with unfortunate death of all patients in the first decade of life. Clinical features include dwarfism, coarse facial features (Fig. 1), severe mental retardation, hirsutism, cardiovascular disease, skeletal abnormalities, and hepatomegaly.12 Acute cardiomyopathy has been described as the presenting feature in some infants younger than 1 year of age.13 Respiratory infection and cardiac failure are the usual causes of death.
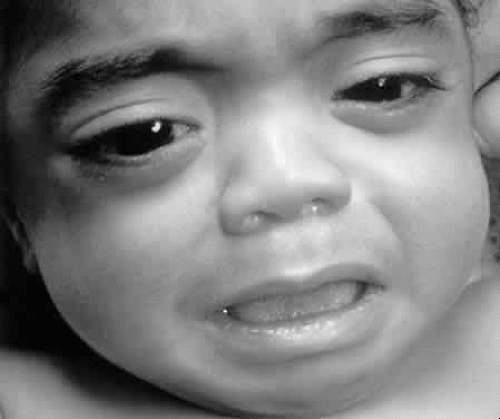 Fig. 1. Coarse facial features of infant with Hurler syndrome (MPS I-H). |
Progressive diffuse punctate stromal corneal opacities occur in all patients. They may conceal retinal degeneration and result in progressive visual loss.14,15 When the retina can be evaluated, a retinitis pigmentosa-like picture indistinguishable from other forms of heredofamilial retinal pigmentary dystrophies usually is present. The electroretinogram is diminished or nonrecordable.16 Glaucoma is relatively rare17 but has been reported in some patients.14,18 Optic nerve head swelling and optic atrophy are common10 (Fig. 2). Congenital cataracts occasionally have been observed.19
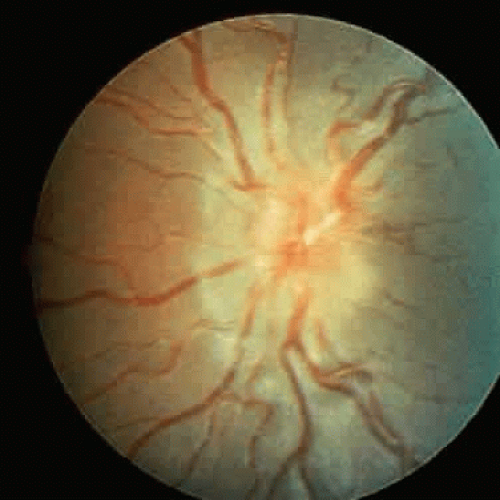 Fig. 2. Optic nerve head and peripapillary elevation in a patient with Hurler syndrome (MPS I-H). |
Hurler syndrome is caused by the absence of α-L-iduronidase, which catalyzes the cleavage of iduronic acid residues from polysaccharide chains. As a result, heparan sulfate and dermatan sulfate accumulate and are excreted in the urine.20 The gene maps to 4p16.3.21 Residual α-L-iduronidase activity in Hurler fibroblasts is heat stable, whereas that in Scheie fibroblasts is heat labile. The enzyme from Hurler-Scheie compound fibroblasts is of inter—mediate activity between Hurler and Scheie-syndromes.
Bone marrow transplantation was performed in two patients homozygous for the relatively common W402X α-L-iduronidase mutation at the ages of 14 and 11 months, respectively. As opposed to untreated patients homozygous for this mutation who have a very severe clinical phenotype with rapid clinical deterioration and death before 6 years of age, the transplanted children were alive at 12 and 14 years of age, respectively. One showed limited mobility but was coping well at school; the patient was wheelchair-bound with severe disability and attended a school for the physically handicapped.22 Transfer and expression of the normal gene in autologous bone marrow may become an alternative method of treatment in the future.23
SCHEIE SYNDROME (MPS I-S; FORMERLY MPS V)
The clinical features of Scheie syndrome24 aremilder than those of Hurler syndrome. Signs and symptoms of the disease usually appear after the age of 5 years, leading to the diagnosis at about 15 years of age. α-L-Iduronidase is deficient, and the disease is allelic with Hurler syndrome.20 Patients have claw-hand deformities, joint stiffness, aortic valve insufficiency, hernias, and deafness. They are of normal height and intelligence and have a relatively normal life span. Facial features are only slightly coarse.
The predominant ocular feature is a peripheral corneal opacification that progresses centrally with age, eventually resulting in visual loss.25 Mucopolysaccharides accumulate in all cellular components of the cornea, with profound alteration of Bowman’s layer and of the corneal lamellae. Ophthalmologic manifestations include glaucoma and pigmentary retinal degeneration that occurs in the first decade of life and is accompanied by night blindness and visual field constriction in the teens and 20s. If vision is reduced markedly, penetrating keratoplasty may be considered even though poor results have been reported, presumably because of the accompanying retinal degeneration.24
As in MPS I-H, dermatan sulfate and heparan sulfate are present in the urine. Scheie syndrome should be differentiated from mild MPS VI-B.
HURLER-SCHEIE COMPOUND (MPS I H-S)
Patients with MPS IH-S have a phenotype of intermediate severity between Hurler and Scheie syndromes.8 The disease is a genetic compound of the two alleles, H and S. Most patients have severe bone involvement, with little or no intellectual impairment. They have a longer life expectancy than those with Hurler syndrome. Micrognathia is a prominent clinical feature and leads to a characteristic facial appearance (Fig. 3). Arachnoid cysts with spinal rhinorrhea are characteristic and may lead to the enlargement of the sella turcica. The cervical cord may be compressed as a result of MPS accumulation in the dura. Symptoms appear at about 5 years of age, and survival to adulthood is common.
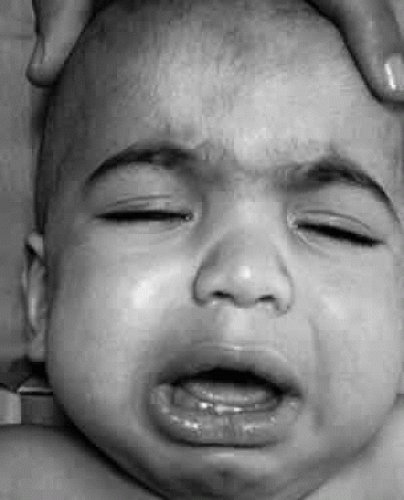 Fig. 3. Facial features of patient with Hurler-Scheie compound (MPS I -HS). |
There is progressive corneal clouding, chronic disc edema, retinal degeneration, and a diminished or extinguished electroretinogram. When there is advanced vision loss, penetrating keratoplasty can be very helpful. Glaucoma is rare. Biochemical and enzymatic assays are identical to those in MPS I-H and I-S.
The residual α-L-iduronidase activity in Hurler fibroblasts is heat stable; it is labile in Scheie fibroblasts. The enzyme activity in MPS IH-S is intermediate between the two. Four novel mutations underlying mild or intermediate forms of α-L-iduronidase deficiency have been discovered, providing insight into genotype-phenotype correlation in this group of patients.26
HUNTER SYNDROME (MPS II)
There are mild (type B) and severe (type A) forms of Hunter syndrome. They are distinguished on clinical grounds only and have a wide spectrum of clinical severity. The two types are allelic and are caused by mutations at the X-linked locus for the enzyme iduronate sulfate sulfatase. Wilson et al27 localized the gene to Xq28, distal to the fragile X site. Characteristic pebbly, ivory-colored skin lesions over the back, neck, scapula, and thigh are present in patients with MPS II (Fig. 4).
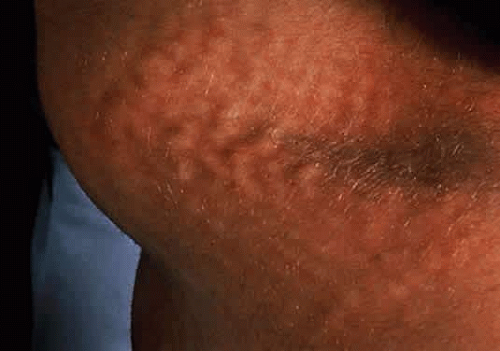 Fig. 4. Nodular skin lesions in the scapular area of a patient with Hunter syndrome. |
Individuals with the severe form have many features of Hurler syndrome, but of lesser severity and with slower progression of somatic and CNS disease. Only males are affected. Death usually occurs before the age of 15 years.
In the milder type B, survival may extend past the age of 45 years.28 The longest known survival is to 87 years of age.29 Intelligence is normal and skeletal involvement moderate, with minimal or no CNS involvement. Corneal clouding usually is absent in type B,28 although deposits of acid mucopolysaccharides are found histologically, even in clinically clear corneas.30 Pigmentary retinopathy with an abnormal or extinguished electroretinogram may lead to visual impairment of varying degrees in all patients.31 In patients with mild Hunter syndrome without raised intracranial pressure, elevated and blurred disc margins lead to the diagnosis of chronic-papilledema, which may culminate into optic-atrophy.10,28,32,33
Dermatan sulfate and heparan sulfate are present in the urine. Definitive diagnosis is made by assaying for the activity of sulfoiduronate sulfatase in fibroblasts. The enzyme defect can be corrected in vitro using an amphotropic retroviral vector containing the human gene.34
SANFILIPPO SYNDROME (MPS III)
Patients with this autosomal recessive syndrome have severe CNS degeneration but only mild somatic disease. Onset of clinical features occurs between 2 and 6 years of age. Significant delay in diagnosis is caused by the mild somatic (Fig. 5) and radiographic features. There may be moderate dwarfism, minimal skeletal dysostosis, and moderate hepatosplenomegaly. The presenting problem may be that of marked hyperactivity, destructive tendencies, and other behavioral aberrations. Four biochemically distinct types of MPS III are difficult to differentiate clinically. Type A is most severe, with an earlier onset, more rapid progression of symptoms, and earlier death than types B, C, or D.35 Types B and D are heterogeneous.36,37. Type C is intermediate between A and B. Excessive heparan sulfate, but not dermatan sulfate, is excreted in the urine. N-sulfated glucosamine residues are removed during the degradation of heparan sulfate through the sequential action of four enzymes that are defective in Sanfilippo syndrome. In Type A, there is a deficiency of heparan N-sulfatase or sulfamidase; in type B, α-N-acetyl-glucosaminidase (NAG) is lacking; in type C, acetyl CoA: α-glucosaminidase is deficient; and in type D, the defect is in N-acetyl glucosamine 6-sulfatase.
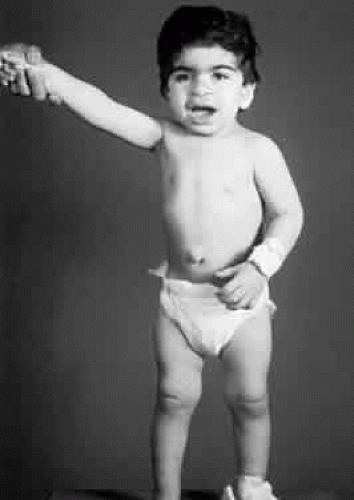 Fig. 5. Relatively normal facial features in a patient with Sanfilippo syndrome type A (MPS IIIA). |
The corneas are clear clinically but show similar but less severe ultrastructural abnormalities as other forms of MPS.15 A retinitis pigmentosa-like fundus picture frequently is present.38 Optic atrophy may be present.
MORQUIO SYNDROME (MPS IV)
Two types of Morquio syndrome are recognized with a wide spectrum of clinical manifestations. Type A is caused by deficiency of N-acetyl galactosamine 6-sulfatase,42 and type B is caused by deficiency of β-galactosidase.43 As with most MPSs, no clinical abnormalities are apparent at birth. An awkward gait, retarded growth, knock knees, sternal bulging, and flaring of the rib cage become evident in the second year of life. Patients with Morquio syndrome have short trunk dwarfism (Fig. 6). They are of normal intelligence, and their distinctive skeletal abnormalities become prominent in the first 10 years of life. The predominant clinical features are related to the effect of the disease process on the spinal cord.44 The joints are loose and hyperextensible, the wrists enlarged, and the hands misshapen,8 and there is genu valgum and kyphosis. The facies is characteristic with a broad mouth, prominent maxilla, short nose, and widely spaced teeth.7 Odontoid hypoplasia may lead to atlantoaxial subluxation and spinal cord compression later in the course of the disease. Cervical myelopathy develops early. Death occurs late in childhood from respiratory paralysis secondary to spinal cord compression or from recurrent pneumonia.
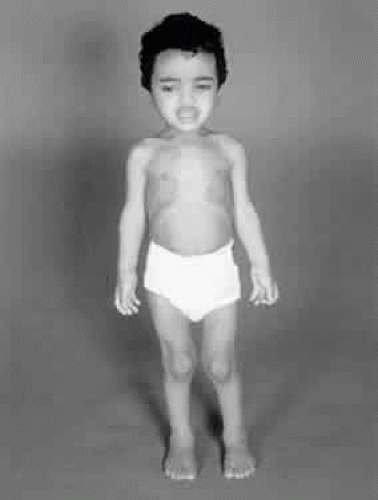 Fig. 6. Child with Morquio syndrome (MPS IV). |
Corneal clouding, the most common ocular feature, generally is not present before 10 years of age45 (Fig. 7), but one of us (EIT) has observed it in three Lebanese siblings younger than 10 years of age. The corneal epithelium and Bowman’s layer appear normal under the slit lamp, and there is a homogeneous clouding of the corneal stroma. There is no retinal dystrophy.46
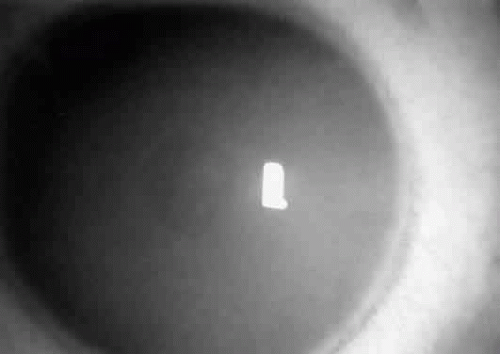 Fig. 7. Corneal opacification in a child with Morquio syndrome (MPS IV). |
The gene for type A maps to 16q24.3.47
MAROTEAUX-LAMY SYNDROME (MPS VI)
This autosomal recessive disease was first recognized in 1963.48 Severe and mild phenotypes have been described.7 Type A, or classic form, is more severe than its allelic form, type B. The deficient enzyme is arylsulfatase B (N-acetylgalactosamine 4-sulfatase).49 Heparan sulfate and dermatan sulfate are excreted excessively in the urine. The extent of corneal clouding is the same in both types, but skeletal dysplasia is less pronounced in the latter. The clinical features, except for normal intelligence, are reminiscent of those of Hurler syndrome, especially in the severe form (Fig. 8).
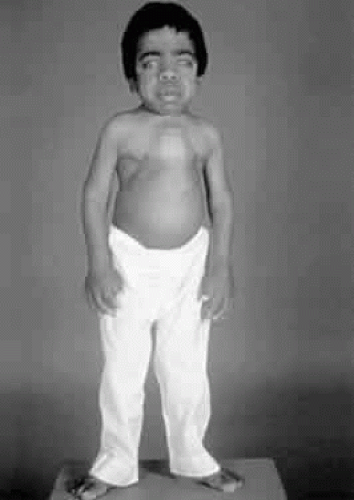 Fig. 8. Facial and skeletal abnormalities in a child with Maroteaux-Lamy syndrome (MPS VI). |
Growth retardation is first noted at 2 or 3 years of age. Restriction of joint movement develops in the first year of life. Significant cardiomyopathy may arise, and hepatosplenomegaly may be present. Hypoplasia of the odontoid process can cause spinal cord compression and spastic paraplegia.50 Umbilical and inguinal hernias are common. Patients with type A Maroteaux-Lamy syndrome die in their teens from hydrocephalus secondary to meningeal infiltration with mucopolysaccharides. Radiographic findings are similar to those of patients with Hurler syndrome and are striking examples of dysostosis multiplex.
Diffuse corneal stromal opacification develops early in life. The cornea increases in thickness, especially in its periphery, where the clouding is most dense.11 Papilledema and optic atrophy occur.10,51 Retinal degeneration with electroretinographic abnormalities is a feature of the disease. Corneal transplantation has been performed in patients with-Maroteaux-Lamy syndrome, but the long-term visual outcome is poor.52,53 Vision may be poor despite clear corneal grafts because of associated retinal or optic nerve disease.
The gene maps to 5q11–q13.54
SLY SYNDROME (MPS VII)
β-Glucuronidase is deficient in this type of mucopolysaccharidosis.55 There may be two allelic forms leading to two clinical phenotypes with a wide range of severity of signs and symptoms.
The gene map for β-glucuronidase was refined to 7q21.11 by Ward et al56 using dosage analysis of chromosomal aberrations. There is excessive urinary excretion of glycosaminoglycans and coarse granulocyte inclusions in a variety of tissues.
The severe form of the disease is characterized by rapidly progressive mental, motor, and growth retardation. Patients have hepatosplenomegaly, massive ascites, inguinal hernias, thoracolumbar gibbus, and skeletal radiographic changes similar to those of the other mucopolysaccharidoses. Individuals with the milder phenotype exhibit hepatosplenomegaly, skeletal abnormalities, and an unusual facies. Mental retardation is not present at birth but develops with aging. Corneal clouding is variable and usually mild.57 A mild pigmentary retinopathy develops later in life.
DISORDERS OF LYSOSOMAL ENZYME PHOSPHORYLATION
Spranger and Wiedemann coined the term mucolipidoses for this group of disorders.58 The mucolipidoses have phenotypic and biochemical features of both the mucopolysaccharidoses and the sphingolipidoses without excessive urinary excretion of mucopolysaccharides. Ultrastructurally, the affected cells resemble those of the MPSs, with cytoplasmic vacuoles filled with fine fibrillogranular-material. The vacuoles also contain membranous lamellar inclusions similar to those present in the lipidoses.15
Four diseases have been included in this category: 1) mucolipidosis I, or sialidosis; 2) mucolipidosis II, or inclusion cell disease (I-cell disease); 3) mucolipidosis III, or pseudo-Hurler polydystrophy; and 4) mucolipidosis IV, or Berman disease.
MUCOLIPIDOSIS I (MLS I) (SIALIDOSIS TYPE 1; CHERRY-RED SPOT MYOCLONUS SYNDROME)
In MLS I, there is deficiency of the enzyme sialidase that leads to a syndrome characterized by a retinal cherry-red spot and myoclonus. MLS I was reclassified as sialidosis type 159 and is discussed later in this chapter.
MUCOLIPIDOSIS II (MLS II) (I-CELL DISEASE)
Patients with this rare and severe autosomal recessive mucolipidosis display many of the clinical and radiographic features of Hurler syndrome.60 There are striking fibroblast inclusions, hence the name I-cell disease.61 There is no mucopolysacchariduria.62,63 The disease results from the absence of the enzyme that attaches a recognition phosphate group onto a mannose residue in hydrolases. There is abnormal lysosomal enzyme transport in cells of mesenchymal origin. Severe progressive psychomotor retardation occurs. MLS II is differentiated from Hurler syndrome by the earlier onset of signs and symptoms, the absence of mucopolysacchariduria, the early and striking gingival hyperplasia, and the rapidly progressive course leading to death in the first decade.64
Early in development, congenital dislocation of the hips, thoracic deformities, hernia, and gingival hyperplasia are evident.65,66 Radiographic studiesshow bony changes of dysostosis multiplex more severe than in MPS I. Patients have coarse facial features, craniofacial abnormalities, and restricted joint mobility.
Early in the course of the disease, the corneas are clear. Late corneal clouding is common; it correlates positively with survival and occurs in 40% of cases.67 Corneal opacities are evident on slit-lamp examination as diffuse stromal granules. Glaucoma occurs in 6% of patients.68 No cherry-red macula is present. Conjunctival biopsy is diagnostic.68
The diagnosis can be made biochemically by demonstration of elevated serum and urine lysosomal enzyme levels and by measuring the UDP-N-acetylglucosamine lysosomal enzyme N-acetylglucosamine phosphotransferase (GNPTA) in fibroblasts. The gene for this enzyme maps to 4q21–q23.69 Prenatal diagnosis is reliable and carrier detection is possible. There is no definitive treatment.
MUCOLIPIDOSIS III (MLS III) (PSEUDO-HURLER POLYDYSTROPHY)
Mucolipidosis III results from the same enzyme deficiency as MLS II. N-acetylglucosamine 1-phosphotransferase is composed of at least two distinct polypeptides: a recognition subunit that is defective in the MLS III variant and a catalytic subunit that is deficient or altered in the classic forms of MLS II and III as well as in the MLS II variant.70
Onset of clinical signs and symptoms is later than in MLS II. The clinical course progresses more slowly, and survival into adulthood is possible.71,72 Most patients have been Ashkenazi Jews. Children usually present at about 3 years of age with joint stiffness, coarse facial features, and short stature.73 They have many of the features of Hurler syndrome but with a much slower clinical evolution and no mucopolysacchariduria. The arms and hands are involved most markedly, and carpal tunnel syndrome with wasting of the thenar eminence is common (Fig. 9). Aortic valve disease occurs in most cases. The pathology of MLS III is not well documented. Cultured fibroblasts have inclusion bodies similar to, but not as prominent as, MLS II cells.
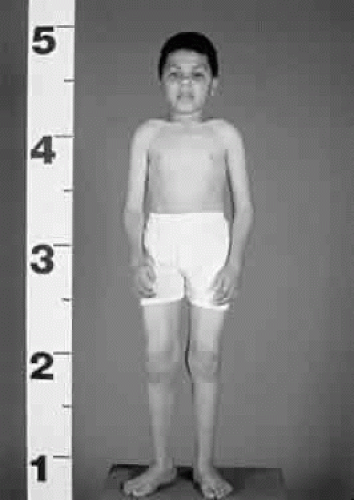 Fig. 9. Child with mucolipidosis III. Note mild coarse facial features and joint contractures, most prominent in the hands. |
Nearly 50% of reported patients have some learning disability or mental retardation.74 Life expectancy and the natural course of the illness beyond the third decade are unknown.
Corneal clouding (Fig. 10), hyperopic astigmatism, and a mild retinopathy with surface-wrinkling maculopathy (Fig. 11) appear to be the constant ocular triad.75 Some patients have retinal vascular tortuosity, optic nerve head swelling (Fig. 12), visual field defects, and abnormalities in color vision. Visual complaints are uncommon.
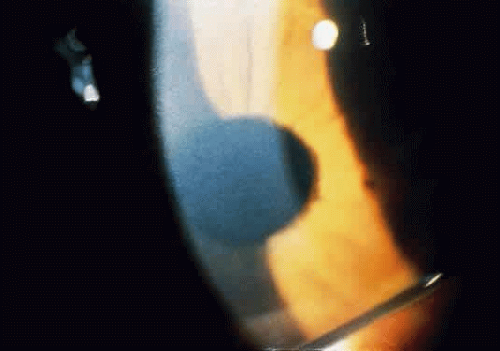 Fig. 10. Mild corneal clouding in a child with mucolipidosis III. (Traboulsi E, Maumenee I: Ophthalmologic findings in mucolipidosis III. Am J Ophthalmol 102:529, 1986) |
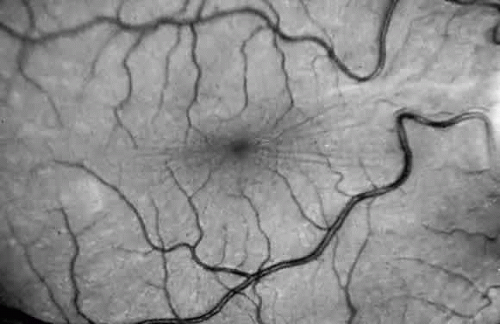 Fig. 11. Surface-wrinkling maculopathy in a patient with mucolipidosis III. (Traboulsi E, Maumenee I: Ophthalmologic findings in mucolipidosis III. Am J Ophthalmol 102:529, 1986) |
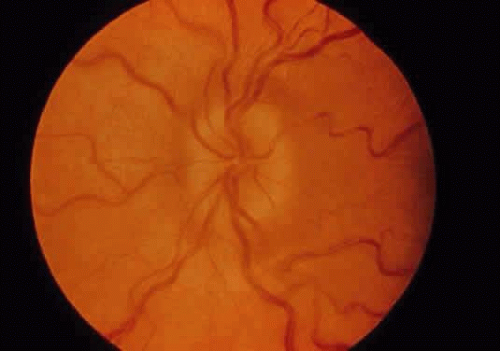 Fig. 12. Optic nerve head swelling in a patient with mucolipidosis III. (Traboulsi E, Maumenee I: Ophthalmologic findings in mucolipidosis III. Am J Ophthalmol 102:529, 1986) |
Prenatal diagnosis by means of amniocentesis is possible. There is no specific or definitive treatment.
MUCOLIPIDOSIS IV (MLS IV) (BERMAN DISEASE)
Mucolipidosis IV was first described by Berman.12 It is characterized by psychomotor retardation, lack of skeletal deformities and organomegaly, and variable ophthalmologic abnormalities in the form of corneal opacities and retinal degeneration. Patients have been described with onset of clinical findings from 1 year of age to the mid 20s. Life span and prognosis beyond this age remain to be determined. The disease is seen mainly among Ashkenazi Jews.76 MLS IV differs from most other lysosomal storage disorders in the apparent lack of progression in some patients. In most, however, there is marked psychomotor and physical retardation. Biochemically, MLS IV is characterized by the accumulation of gangliosides,77 phospholipids,78 and acidic mucopolysaccharides.79 Deficiency of ganglioside sialidase was reported as the possible metabolic defect causing this disorder, and partial reduction in activity of this enzyme was noted in obligate heterozygotes.80 Conjunctival biopsy shows characteristic-intracellular inclusions.81
There is progressive severe visual impairment from corneal opacification12,82 and from progressive retinal degeneration.83,84 The age of appearance and the extent and severity of the clinical course of these abnormalities are variable. The superficial corneal opacification is a characteristic feature of MLS IV and leads to the clinical diagnosis in most children. The opacities are not congenital in all patients, as suggested previously.12 With time, the corneal opacities may increase in density, with poor fundus visualization leading to the erroneous diagnosis of cataracts in some patients. Epithelial edema also hasbeen noted. Corneal clearing may occur after epithelial removal, but clouding returns after a few days or weeks. A progressive rod-cone dystrophy is present with a subnormal or extinguished electroretinogram (ERG).82,84 The static nature and, at times, improvement in the degree of corneal opacification in more than 50% of cases is characteristic of MLS IV. In other lysosomal storage disorders, corneal clouding and visual impairment are progressive. Deterioration of visual function in MLS IV correlates with retinal degeneration rather than with worsening of corneal opacities. Prenatal diagnosis is possible.81,82
DISORDERS OF GLYCOPROTEIN DEGRADATION AND STRUCTURE
Glycoproteins are synthesized via two pathways: (1) the glycosyltransferase pathway that synthesizes oligosaccharides linked to serine or threonine and (2) the dolichol, lipid-linked pathway that synthesizes oligosaccharides linked to asparagine. Specific enzyme deficiencies cause individual glycoprotein storage diseases.
MANNOSIDOSIS
There are α and β types of mannosidosis.85 We restrict our comments to α-mannosidosis because of the associated ophthalmologic abnormalities. α-mannosidosis results from a deficiency of the lysosomal enzyme α-mannosidase. There are elevated serum levels and excretion of small mannose-richoligosaccharides. Patients with α-mannosidosis excrete a unique pattern of increased amounts of several oligosaccharides.86,87 The gene for α-mannosidase was mapped to chromosome 19p13.2-q12.88 In severe infantile type I mannosidosis, there is rapidly progressive mental retardation, hepatosplenomegaly, severe dysostosis multiplex, and often death between the ages of 3 and 10 years.89 The milder juvenile and adult type II mannosidosis is characterized by a more slowly progressive course with survival into adulthood.90 Hearing loss is a prominent feature of this disease, but clinical heterogeneity is evident.91 Recurrent bacterial infections and hernias are seen.92,93 Types I and II are not clearly separated. Ophthalmologic manifestations are present in both types and consist of superficial corneal opacities and posterior spoke-like lens opacities.94
FUCOSIDOSIS
Fucosidosis is caused by deficiency of the lysosomal enzyme α-fucosidase.97 This results in accumulation and excretion of a variety of glycoproteins, glycolipids, and oligosaccharides containing fucoside moieties. The disorder is panethnic, with a higher incidence in Italy and in the Southwestern parts of the United States.98 There is faulty degradation of both sphingolipids and polysaccharides.99 Urine samples from individuals with fucosidosis contain excessive amounts of several fucoglycoconjugates.100 The most precise way of diagnosing fucosidosis is based on enzymatic assay of α-L-fucosidase in cells of any type.
There is a spectrum of severity of the clinical findings with mild to severe phenotypes. The more severely affected patients exhibit psychomotor retardation, coarse facies, growth retardation, dysostosis multiplex, and neurologic retardation in the first year of life. The milder phenotype is characterized by angiokeratomas (Fig. 13) and by a longer survival. The gene for fucosidosis maps to chromosome 1p24.101
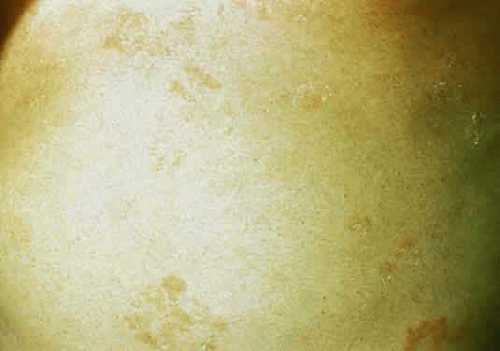 Fig. 13. Cutaneous lesions in fucosidosis. |
SIALIDOSIS
Sialidosis is a rare lysosomal storage disease with clinical features resembling those of Hurler syn-drome, but milder. The two forms of the disease, sialidosis type 1 and type 2, result from deficiency of neuraminidase. The gene defects of the two types are not allelic. Complementation studies between sialidosis type 1 and type 2 fibroblasts result in restoration of both β-galactosidase and sialidase activities in fused cells. Moderate but progressive mental retardation, cerebellar signs, peripheral neuropathy, myoclonic jerks, tremor, dysostosis multiplex, and peculiar inclusions are present in cultured fibroblasts,8,105,106 but there is no mucopolysacchariduria. There is accumulation of syalyloligosaccharides in various tissues and organs of the body and deficiency of the enzyme α-N-acetylneuraminidase in cultured fibroblasts.107
Diagnosis is based on electron microscopy. Fibroblasts in conjunctival biopsy specimens contain small membrane-bound vacuoles containing fibrillogranular and membranous lamellar bodies detectable by electron microscopy.15 The differential-diagnosis must include other diseases causing a cherry-red macula. There is no known treatment, but prenatal diagnosis is possible.
Sialidosis Type 1
Sialidosis type 1 is characterized by the cherry-red spot myoclonus phenotype (cherry-red spot myo-clonus syndrome) and is the milder form of sialidosis. There are at least 15 confirmed patients.108,109 The disease results from a defect in the structural gene for sialidase, which maps to 10pter-q23.110 Retinopathy and myoclonus occur simultaneously at the onset of the disease, which tends to be in early adolescence but can be variable. The striking neurologic manifestation is a stimulus-sensitive myoclonus that limits daily activities. Ataxia and generalized grand-mal seizures also may occur. Intellect is preserved, and patients with this rare disorder can survive beyond 30 years of age.
The presenting symptom may be a reduction in visual acuity; visual loss is progressive and often severe. It may be associated with impaired color vision and night blindness.111 The macular cherry-red spot is consistent but can be atypical. Punctate lens opacities can occur.109,112,113 Nystagmus, optic-atrophy, and visual field constriction have been-described.
An important diagnostic screening test in this disorder is the examination of the urine for excessive excretion of sialic acid-containing compounds. Urine thin-layer chromatography stained with resorcinol gives a blue color, suggesting the presence of sialylated oligosaccharides. The definitive test is the demonstration of deficient sialidase in fresh cultured skin fibroblasts.
Sialidosis Type 2
Sialidosis type 2 is distinguished from sialidosis type 1 by the early onset of a severe progressive muco-polysaccharidosis-like phenotype with visceromegaly, dysostosis multiplex, and mental retardation. The disease may be the result of a defect in the gene for a protective protein on chromosome 20.110 There are several subtypes with marked variability in clinical manifestations. Recognized are a congenital, or hydropic form, an infantile form, and a juvenile form, the latter of which is the most common and which is described in this chapter under the title of galactosialidosis.
Congenital sialidosis type 2 patients all have been stillborn infants with hydrops fetalis, ascites, hepatosplenomegaly, stippling of the epiphyses, and periosteal cloaking of the long bones. There are no documented ophthalmologic findings.
Patients with infantile-onset sialidosis type 2 are relatively healthy at birth.114 Later, a progressive severe MPS-like phenotype develops. Development is slow, with mental retardation and gait ataxia. Myoclonus is present. Grandmal seizures, deafness, and a peripheral neuropathy may occur. Skeletal abnormalities are prominent with dysostosis multiplex. Vision is retained despite the presence of a macular cherry-red spot and punctate lens opacities.
Peripheral blood lymphocytes are vacuolated, and foam cells are present in the bone marrow. Sialidase is deficient in cultured fibroblasts.
Juvenile sialidosis type 2 is described under β-galactosialidosis.
ASPARTYLGLUCOSAMINURIA
Aspartylglucosaminuria is an autosomal recessive disorder that occurs primarily in Finland.115 Rare, isolated cases are reported from other countries. The accumulation of aspartylglucosamine results from a deficiency of the lysosomal enzyme, aspar—tylglucosaminidase, which maps to chromosome 4q32–q33.116
Patients are healthy for the first few months of life. Recurrent infections, diarrhea, and hernias are noted during the first year of life. Coarsening of facial features and saggy skin folds occur in the first decade. Mental deterioration begins between the ages of 6 and 15 years. Crystal-like lens opacities are observed.117 Aspartylglucosamine is the major storage compound found in tissues and fluids of patients with this disease.118 There may be vacuolated lymphocytes and neutropenia. The urine contains large amounts of aspartylglucosamine. No treatment is known.
SCHINDLER DISEASE
Schindler disease is caused by deficient activity of the lysosomal hydrolase enzymes α-N-acetylgalactosaminidase and α-galactosidase B.119 The gene has been localized to 22q13.1–q13.2.120 The disease is clinically heterogeneous, and there are two major phenotypes. Only type 1 is described because of its ophthalmologic features.
In type 1 Schindler disease, there is infantile onset of severe neural degeneration and neuroaxonal dystrophy. The disorder originally was described in two German brothers who were the products of a consanguineous marriage.119,121 Development is normal in the first 9 to 15 months of life. This is followed by rapid neural degeneration and by loss of developmental milestones, resulting in severe psychomotor retardation, myoclonic seizures, and cortical blindness. Limited information is available on pathologic and biochemical abnormalities because all known patients are still living.
Ophthalmologic findings include strabismus, optic atrophy, nystagmus, and eventually, cortical blindness. Neuroimaging studies demonstrate generalized atrophy of the brainstem, cerebellum, and cortex. Visual evoked potentials have low amplitude, delayed responses, or both.121,122
Diagnosis of affected homozygote and heterozygote carriers can be made only by determination of α-N-acetylgalactosaminidase activity in various tissues. Prenatal diagnosis is possible. There is no specific treatment, and appropriate supportive care should be implemented as needed.
DISORDERS OF LIPID METABOLISM
NIEMANN-PICK DISEASE
Niemann-Pick disease (NPD) results from impaired sphingomyelin metabolism. The disease was first reported by Niemann in 1914.123 Thirteen years later, Pick described the characteristic vacuolated or foam cells in many body tissues.124 NPD results from deficient activity of acid sphingomyelinase (ASM) that maps to 18q11–q12.125
Crocker126 categorized the NPD phenotypes into four clinical entities designated A to D. The broad classification of NPD into five phenotypic variants (A through E) followed and was based on the age of onset, the severity and type of neurologic involvement, and the evolution of the disease. Hepatosplenomegaly and foam cells in the bone marrow are constant features in all variants. Type A is the infantile neurodegenerative form. Type B is distinguished from the severe neuronopathic type A by the absence of neurologic involvement, by the presence of hepatosplenomegaly, and by survival into adulthood. Patients with type C NPD are healthy for the first 1 or 2 years of life and then experience a slowly progressive and variable neurodegenerative course, with less severe hepatosplenomegaly and survival into adulthood; they often present with prolonged neonatal jaundice. Type D patients share a common ancestry traceable to Acadians from Nova Scotia. Neurologic symptoms develop later in childhood, and these patients have a slower neurodegenerative course than those with type C. Type E is the adult form with a mild degree of visceral sphingomyelin storage, mild splenomegaly, foam cells in the bone marrow and absence of neurologic signs.
ASM is deficient in types A and B, which are believed to be allelic.127 In type A NPD, ASM is reduced dramatically,128,129 whereas in type B, there is residual ASM activity.130 In type C, sphingomyelinase activity usually is normal.131 The metabolic block in type C is in the intracellular trafficking of cholesterol.132 The biochemical defects in type D and type E NPD remain to be determined.
The pathologic hallmark of types A and B is the Niemann-Pick cell, which is a lipid-laden foam cell133 (Fig. 14). Sphingomyelin accumulates in the brain and autonomic ganglia. The neurons become swollen and have a pale cytoplasm. Ultrastructurally, the cells contain concentric lamellated bodies representing storage cytosomes. Inclusion profiles in the viscera, lymph nodes, and foam cells also correlate with an increase in sphingomyelin content. Diagnosis can be made readily by enzymatic determination of ASM activity in cells and tissues. More than 300 cases of type A and B NPD have been reported. Prenatal diagnosis has been accomplished by enzyme assays of cultured amniotic fluid cells in types A and B.
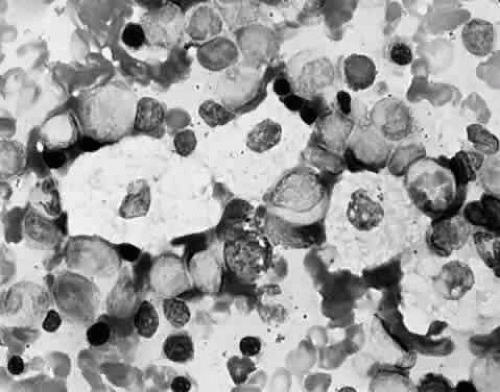 Fig. 14. Foam cells in bone marrow aspirate of patient with Niemann-Pick type B. Giemsa stain. |
Niemann-Pick Disease Type A
Type A NPD is fatal in infancy. It is characterized by failure to thrive, hepatosplenomegaly, and severe CNS involvement, with loss of motor function leading to death by 2 to 3 years of age. A higher incidence is seen in the offspring of Ashkenazi Jews compared with the general population.134
A cherry-red macula is present in 50% of infants in the first and second year of life.135 There is no distinction between the appearance of the cherry-red spot in infantile NPD and in Tay-Sachs disease.136,137 Occasionally, a macular halo syndrome with a gray, granular-appearing macula is observed.138 Optic atrophy develops with time. Subtle lens opacities and corneal clouding can occur.137 ERG is abnormal. The stored lipid is localized to the ganglion and amacrine cells of the retina, most conspicuously in the parafoveal region. The other retinal layers appear unaffected.135
Niemann-Pick Disease Type B
Type B NPD has a variable phenotype, with only visceral involvement. It is diagnosed in childhood between 3 and 11 years of age, or in adult life with hepatosplenomegaly and progressive pulmonary infiltrates that cause the major disease complications.139 Most patients have a normal intellect and survive into adulthood. Patients with type B NPD are of mixed ethnic backgrounds. Harzer and associates140 were first to demonstrate a low sphingomyelinase level in this disease.
A unique retinal abnormality, the macular halo syndrome, has been reported in type B NPD by Cogan and Kuwabara135 and consists of a ring of opacities around the macula that causes no visual impairment (Fig. 15). This abnormality has been reported by several authors.141,142,143 The crystalloid halo which measures about half the disc diameter occurs at the outer edge of the retina mainly in Henle’s fiber layer causing only minor obstruction of the overlying vessels. Matthews and associates144 proposed that the macular halo represents the mildest form of a cherry-red spot in the ganglion cell layer of the retina. Their findings are in conflict with those of Cogan et al.138 The precise location of the opacities in the retina remains uncertain because of the lack of histopathology. The available clinical data suggest that such opacities are permanent.
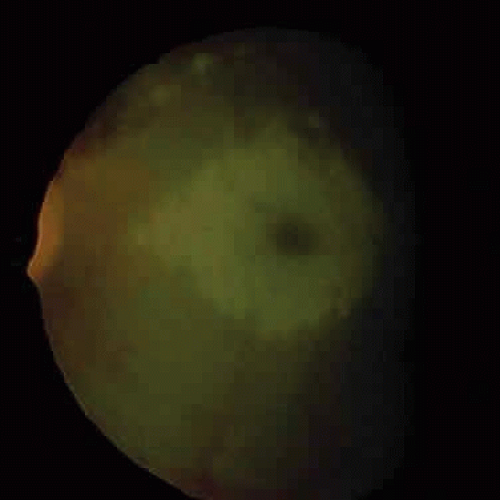 Fig. 15. Macular halo in a patient with Niemann-Pick type B. |
Bone marrow transplantation has been successful in type B NPD. Enzyme replacement and somatic gene therapy using macrophage-targeted recombinant enzyme may be available in the future.
Niemann-Pick Disease Type C
Type C NPD is a panethnic autosomal recessive lipidosis associated with lysosomal accumulation of unesterified cholesterol. It was first recognized in 1958145 and is biochemically distinct from type A and type B NPD.146 It is as common as types A and type B NPD combined. This distinction was supported by somatic cell hybridization studies.147 Genetic isolates have been described with the Nova Scotia form, formerly type D NPD,126 and in southern Colorado.148 There is no evidence of genetic heterogeneity. Sea-blue histiocytes in the bone marrow are common but nonspecific.149 The metabolic block results from defective intracellular cholesterol trafficking.132 The disease is diagnosed by filipin staining and by demonstration of fluorescence around the nucleus after low-density lipoprotein (LDL)-cholesterol loading.150 Cultured fibroblasts from patients and carriers have high levels of unesterified cholesterol. There is no macular cherry-red spot or a macular halo in type C NPD. The differentiating clinical features of type C NPD (ophthalmoplegic neurovisceral lipidosis, vertical supra’s disease) are its three main clinical features, which led Cogan et al151 to recommend the acronym DAF syndrome to denote downgaze paralysis, ataxic athetosis, and foam cells in the spleen, liver, and bone marrow. There is extensive infiltration of bone marrow, spleen, liver, and other tissues with foam cells. Sphingomyelinase activity in leukocytes and cultured fibroblasts is decreased or normal.152,153
Type C NPD has a subacute clinical course and presents in infancy with neonatal hepatitis or later in childhood with moderate splenomegaly and gradual neurologic deterioration. Most patients have seizures and limitation of vertical gaze. The presence of downgaze paresis is characteristic154 and has been noted in all juvenile and adult cases.155 Subtle slowing of vertical saccades begins in late infancy. Voluntary vertical gaze is completely paralyzed in late stages of the illness.156 Horizontal eye movements may be affected with a total supranuclear ophthalmoplegia. Patients usually die before the end of the second decade.
Niemann-Pick Disease Types D, E, and F
Type D NPD is caused by a defect in cholesterol metabolism, and although few biochemical studies have been published, it may be allelic with type C.157,158 The underlying metabolic defect in type D and type E NPD still is not understood, but these two variants cannot be diagnosed by a single laboratory test. Increase in organ sphingomyelin content or presence of foam cells in the bone marrow helps to corroborate clinical suspicions. Neurologic symptoms develop later in childhood in patients with type D disease, and they have a slower neurodegenerative course than type C patients.159 They share a common ancestry traceable to Acadians. There are no ocular abnormalities in type D. Patients with the type E variant may have a macular cherry-red spot. In 1978, Schneider et al160 described two patients with a heat-labile form of ASM and classified it as type F NPD.
FABRY’S DISEASE (ANGIOKERATOMA CORPORIS DIFFUSUM UNIVERSALE)
Fabry’s disease results from a defect in glycosphingolipid catabolism. The disease has an incidence of approximately 1 in 40,000.122 The gene is X-linked recessive and has been localized to Xq22.161 It codes for the lysosomal hydrolase α-galactosidase A.162 Most heterozygous female carriers have an intermediate level of enzymatic activity.163,164 There is progressive and systematic accumulation of the glycosphingolipid ceramide trihexaside, particularly in vascular endothelial cells.165 Carrier females in a pedigree can be examined clinically and biochemically for heterozygote identification.
The disease usually has its onset during childhood or adolescence. Classically, affected hemizygotes have pain and paresthesia of the extremities (acroparesthesia) around the time of puberty166; vascular cutaneous lesions (angiokeratomas) of the scalp, mucous membranes, skin, and inguinal and umbilical regions; hypohidrosis; and the characteristic corneal and lens opacities. Severe renal impairment leads to hypertension and uremia. Death occurs from renal failure or from cardiac or cerebrovascular disease.167
The ocular deposition of glycosphingolipids results in unique and diagnostic eye findings in severely affected hemizygous males and minimally affected heterozygous carrier females.168 The ocular findings have been recognized as one of the distinctive hallmarks of this disease and among its earliest clinical manifestations.169 The corneal opacities appear as whorled streaks from a central vortex and have been called cornea verticillata170,171 (Fig. 16). Bilateral inferior granular anterior capsular or posterior subcapsular lens opacities occur in one third of hemizygous males but rarely in heterozygous females. Mild to severe conjunctival (Fig. 17) and retinal vessel tortuosity are present early in life. Visual acuity is not impaired. However, acute visual loss has occurred in hemizygotes as a result of unilateral central retinal vascular occlusion.171 Other ocular findings include lid edema, myelinated nerve fibers, mild optic atrophy, papilledema, nystagmus, and internuclear ophthalmoplegia.171,172 (Fig. 18). Confirmation of the clinical diagnosis in hemizygotes and heterozygotes requires the demonstration of deficient α-galactosidase A activity in plasma, leukocytes, or tears or increased levels of ceramide trihexaside in plasma or urinary sediment. The diagnosis in female heterozygotes can be established by linkage analysis.163,174 These carriers may have some of the systemic manifestations of the disease, but they usually are milder than in affected males. The most frequent clinical finding in females is the characteristic whorl-like corneal epithelial dystrophy. Corneal opacification in female carriers may be more prominent than in male patients because of the admixture of corneal epithelial cell lines with the normal gene and those with the abnormal gene (lyonization). Abnormal glycosphingolipid deposits are found in all ocular and orbital vessels, in smooth muscle of iris and ciliary body, and in perineural cells and connective tissue of the lids and cornea.175
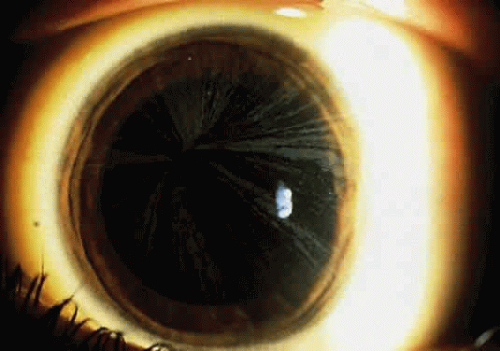 Fig. 16. Cornea verticillata in a carrier female of Fabry disease. (Courtesy of Irene H. Maumenee, MD) |
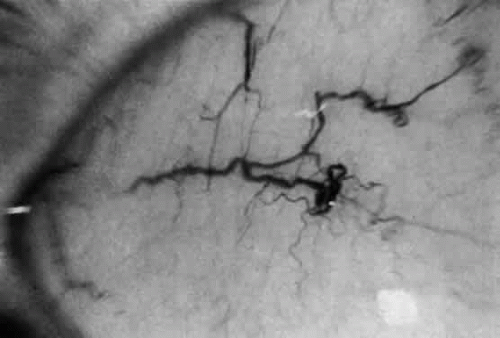 Fig. 17. Corkscrew-like tortuosity and dilatation of conjunctival vessels in Fabry’s disease. (Courtesy of George Spaeth, MD) |
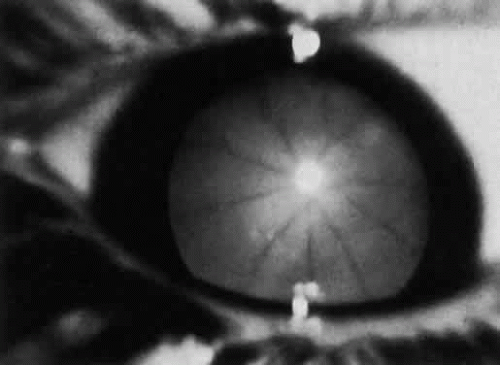 Fig. 18. Spoke-like lens changes in Fabry’s disease. (Courtesy of George Spaeth, MD) |
Prenatal diagnosis is possible by demonstration of the specific α-galactosidase A mutation in chorionic villi or cultured amniotic cells.176
GAUCHER DISEASE
Gaucher disease is an autosomal recessive lysosomal glycolipid storage disorder characterized by the accumulation of glucocerebroside (glucosylceramide) in reticuloendothelial cells.177,178 The gene coding for the deficient enzyme glucocerebrosidase (acid beta-glucosidase) is located on chromosome 1q21.179 The disease is common in the Ashkenazi Jewish population.178 Three phenotypes are recognized based on the absence (type 1) or presence and severity (types 2 and 3) of primary central nervous involvement.180
Type 1 is most common. It is a chronic, non-neuronopathic disorder of adult onset and it accounts for 90% of cases. Splenomegaly, anemia thrombocytopathic, pathologic bone fractures, bleeding episodes, and a yellow skin pigmentatipon and features the disease. The absence of cerebral involvement ditinguishes it from types 2 and 3. Brownish piguecula-like masses containing Gaucher cells are the only significant ocular feature.181 These lesions enlarge and assume a yellow color. The nasal and temporal bulbar conjunctiva are involved with equal frequency in only one fourth of patients. Their presence and significance in this disease has been questioned by Chu et al,182 who did not find any pingueculae in a group of 10 patients.
Type 2 is the acute neuronopathic infantile form. It has an early onset, with severe CNS involvement, failure to thrive, progressive hepatomegaly, splenomegaly, and dysphagia. Later, persistent retroflexion of the head and signs of pseudobulbar palsy develop. Early and late onset varieties have been delineated.183 The classic Gaucher triad consists of trismus, strabismus, and opisthotonus. Oculomotor abnormalities often are the first manifestation of the disease, with strabismus or oculomotor apraxia.183 Most infants with type 2 disease die within the first 2 years of life.184
Type 3 is the subacute, juvenile neuronopathic form, with onset in the teenage years and a chronic course. The severity of type 3 is intermediate between types 1 and 2. The neurologic features are milder and have a later onset. The first symptoms are the result of massive visceral involvement.185 The disease is common in Norrbottnian Swedes. As in type 2, disorders of eye movements are the usual presenting signs.
An additional variant with cardiac valvular calcifications, abnormal eye movements, and corneal opacification that does not interfere with vision is associated with homozygosity for the D409H mutation in the glucocerebrosidase gene.186,187
A tentative diagnosis is based on the detection of Gaucher cells (storage cells) in the bone marrow. Decreased tissue levels of glucocerebroside activity confirm the diagnosis.188 Mutation analysis of the glucocerebrosidase gene also can be performed. The quality of life of patients with Gaucher disease can be improved by a variety of medical and surgical procedures. The clinical abnormalities can be ameliorated and reversed by repeated infusions of modified acid β-glucosidase (ceredase). Cure theoretically is possible using bone marrow transplantation189
METACHROMATIC LEUKODYSTROPHY
Metachromatic leukodystrophy (MLD), known for many years as “diffuse brain sclerosis,” is an autosomal recessive disorder of myelin metabolism.190,191 It is characterized by accumulation of cerebroside sulfate in the CNS and peripheral nerves. Late infantile, juvenile, and adult forms are recognized, based on the age of onset. Heterogeneity exists within each group. The MLD group of diseases also is classified according to the individual biochemical defect. The more common forms are associated with deficiency of arylsulfatase A. The enzyme is absent in all tissues.192,193 This results in abnormal sulfatide metabolism with demyelination and storage of sulfatides in the central and peripheral nervous systems and increased excretion of sulfatides in the urine. The gene for arylsulfatase A maps to 22q13.31-qter.194 The prevention of MLD mainly relies on identifying carriers in known MLD families and on providing genetic counseling, with the possibility of prenatal diagnosis.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


