Chapter 28 Mechanisms of Macular Edema and Therapeutic Approaches
 For additional online content visit http://www.expertconsult.com
For additional online content visit http://www.expertconsult.com
Introduction
The breakdown of the blood–retinal barrier (BRB), modulated via different growth factors, results from a disturbance of the integrity of the tight junctions.1 Starling’s law predicts that macular edema will develop if the hydrostatic pressure gradient between capillary and retinal tissue is increased. That can occur, for example, in the presence of elevated blood pressure, or if the osmotic pressure gradient is decreased by excessive protein accumulation in the extracellular space within the retina.2
Macular edema as A result of various disease mechanisms
Causes of macular edema
The classic pattern of cystoid macular edema (CME) with the petaloid appearance originating from the fluorescein leakage from perifoveal capillaries may be seen in cases of advanced edema of various origins (Fig. 28.1, panel B online). This includes postsurgical CME as well as CME associated with one of the following conditions: diabetes, vascular occlusion, hypertensive retinopathy, epiretinal membranes, intraocular tumors (e.g., melanoma, choroidal hemangioma), intraocular inflammation (e.g., pars planitis), macroaneurysm, retinitis pigmentosa, choroidal neovascularization, and radiation retinopathy.
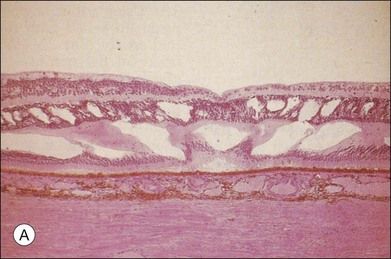
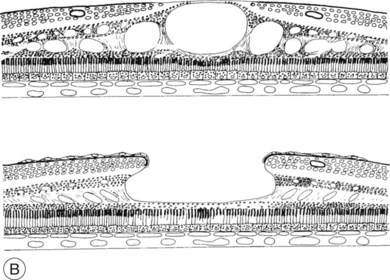
Fig. 28.1, online (B) Schematic drawing of the central retina with large cysts (top), finally resulting in a pseudohole formation (bottom).
Given the heterogeneous etiology of macular edema, its effective treatment depends upon a better understanding of its pathogenesis. In general, formation of macular edema is related to metabolic changes, ischemia, hydrostatic forces, inflammatory and toxic mechanisms, or mechanical forces that occur to various degrees in different conditions (Table 28.1).
Table 28.1 Causes of macular edema in relation to underlying disorders
| Disease group | Disorder | Pathogenesis |
|---|---|---|
| Metabolic alterations | Diabetes | Abnormal glucose metabolism Aldose reductase |
| Retinitis pigmentosa | CME: leakage at the level of RPE | |
| Inherited CME (autosomal-dominant) | Müller cell disease: leakage from perifoveolar capillaries | |
| Ischemia | Vein occlusion Diabetic retinopathy | Inner BRB (retinal capillary hypoperfusion) |
| Severe hypertensive retinopathy HELLP syndrome Vasculitis, collagenosis | Outer BRB (ischemic hypoperfusion of the choroid: serous detachment) | |
| Hydrostatic forces | Retinal vascular occlusions Venous occlusion Arterial hypertension Low IOP | Increased intravascular pressure Failure of the BRB |
| Mechanical forces | Vitreous traction on the macula | Epiretinal membranes with tangential traction Vitreomacular traction syndrome |
| Inflammation | Intermediate uveitis | Mediated by prostaglandins CME is considered an indication for treatment |
| Postoperative CME | Perivascular leukocytic infiltrates | |
| DME | Diabetic leukostasis mediates vascular leakage by endothelial cell apoptosis | |
| Choroidal inflammatory diseases | Vogt–Koyanagi–Harada syndrome Birdshot retinochoroidopathy | |
| Pharmacotoxic effects | e.g., Epinephrine (in aphakia) Betaxolol Latanoprost | Mostly via prostaglandins |
CME, cystoid macular edema; RPE, retinal pigment epithelium; BRB, blood–retinal barrier; HELLP syndrome, hemolytic anemia, elevated liver enzymes, and low platelet count; IOP, intraocular pressure; DME, diabetic macular edema.
Molecular and cellular alterations leading to macular edema
Much of the knowledge on the pathophysiology of macular edema has been determined from extensive experimental studies on diabetic retinopathy and diabetic vascular leakage. A variety of techniques measuring accumulation of material from plasma in the neural retina have been investigated to assess permeability. Such accumulation seems diffuse in nature and focal defects have not been reproducibly described in diabetic mice; as well, interpretations of techniques involving tracer accumulation have not been validated in terms of “gold standard” permeability surface area product.3 Interestingly, edema has not been demonstrated in the retina of diabetic mice based on retinal thickness measurements despite the indication of increased permeability.
The BRB consists of the retinal pigment epithelium (RPE) layer (outer BRB), and the vascular endothelium (inner BRB), that prohibit the passage of macromolecules and circulating cells from the vascular compartment to the extracellular compartment and therefore intraretinal space.4 Intracellular edema (or cytotoxic edema) is defined as cellular swelling that occurs without opening of the BRB. Extracellular (or vasogenic) edema is characterized by retinal thickening in association with loss of BRB integrity (Fig. 28.2). While for diabetes and ischemic retinopathies the inner BRB was found to play a dominant role in vascular leakage, the importance of the outer BRB has recently been supported.5 The outer BRB separates the neural retina from the choroidal vasculature, which is responsible for approximately 80% of the blood supply in the eye. The outer BRB-specific leakage of fluorescent macromolecules can be visualized in diabetic and ischemic rodents and substantial leakage of macromolecules through the outer BRB can be detected.
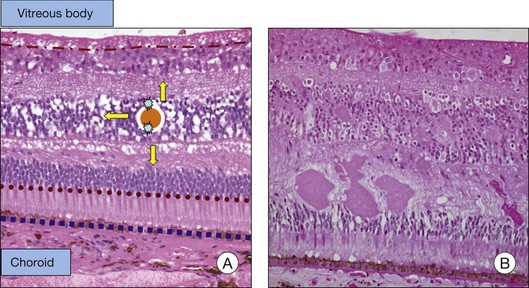
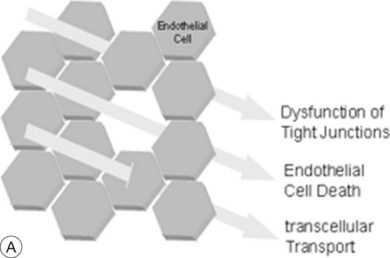
The breakdown of the inner BRB may occur to a variable extent via dysfunction of intercellular junctions, increased transcellular transport, or increased endothelial cell destruction, and result in an increase in vascular permeability (Fig. 28.3B, online).
Cell-to-cell junctions and vascular permeability
Fluid homeostasis and endothelial permeability are mostly regulated by intercellular junctions in the nondiseased retina. Intercellular junctions are complex structures formed by the assembly of a transmembrane and cytoplasmic/cytoskeletal protein components. At least four different types of endothelial junctions have been described: tight junctions, gap junctions, adherence junctions, and syndesmos. Tight junctions are the most apical component of the intercellular cleft (Fig. 28.4, online).
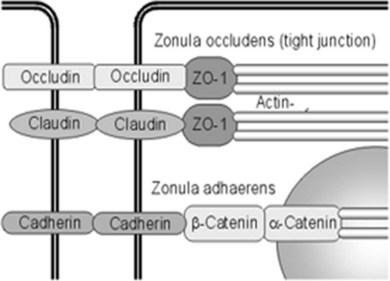
Although the molecular structure of tight junctions generally appears to be similar in all barrier systems, there are some differences between epithelial and endothelial tight junctions, and between tight junctions of peripheral and retinal endothelial cells.6 Expression of selected endothelial cell tight junction genes and particularly that of occludin and claudin-5 is reduced in the diabetic retina.7 In contrast to tight junctions in epithelial systems, structural and functional characteristics of tight junctions in endothelial cells respond promptly to ambient factors. It is likely that inflammatory agents increase permeability by binding to specific receptors that transduce intercellular signals, which in turn cause cytoskeletal reorganization and widening of the interendothelial clefts. For example, tumor necrosis factor-alpha (TNF-α) signals through protein kinase C (PKC)ζ/nuclear factor-kappa B (NF-κB) to alter the tight junction complex and increase retinal endothelial cell permeability.8 Endothelial junctions also regulate leukocyte extravasation. Once leukocytes have adhered to the endothelium, a coordinated opening of interendothelial cell junctions occurs.
Inflammation and vascular permeability
In diabetes, activated leukocytes adhere to the retinal vascular endothelium.9,10 Increased leukostasis is one of the first histologic changes in diabetic retinopathy and occurs prior to any apparent clinical pathology.
Adherent leukocytes play a crucial role in diabetic retinopathy by directly inducing endothelial cell death in capillaries,11 causing vascular obstruction and vascular leakage. Endothelial cell death precedes the formation of acellular capillaries.10 With time, however, acellular capillaries prevail and become widespread. Although the mechanism of this destructive process remains elusive, it is clear that the interaction between the altered leukocytes and the endothelial cells and the subsequent endothelial damage represents a crucial pathogenic step9,11,12 (Fig. 28.5).
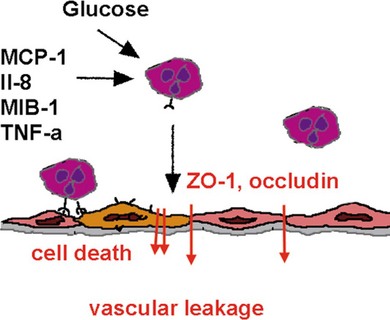
Inflammatory cytokines such as TNF-α decrease the protein and mRNA content of the tight junction proteins zonula occludens (ZO)-1 and claudin-5.8 TNF-α and interleukin-1 beta (IL-1β) are elevated in the vitreous of diabetic patients and in the retina of diabetic rats associated with increased retinal vascular permeability and leukostasis13,14 (Fig. 28.6, online). Furthermore, TNF-α is involved in ischemic vascular changes.15
Growth factors, vasoactive factors, and vascular permeability
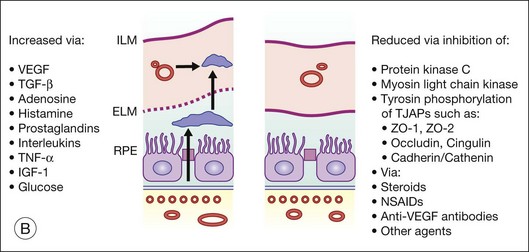
The disruption of endothelial integrity leads to retinal ischemia and vascular endothelial growth factor (VEGF)-mediated iris and retinal neovascularization.9,16,17 VEGF is 50 000 times more potent than histamine in causing vascular permeability.18–20 Previous work has shown that retinal VEGF levels correlate with diabetic BRB breakdown in rodents21 and humans.22 Flt-1(1–3 Ig)Fc, a soluble VEGF receptor, reverses early diabetic BRB breakdown and diabetic leukostasis in a dose-dependent manner.17 Early BRB breakdown localizes, in part, to retinal venules and capillaries of the superficial inner retinal circulation23 and can be sufficiently reduced by VEGF inhibition (Fig. 28.7, online). Although VEGF is only one of the cytokines involved in the pathogenesis of the vascular leakage, it is likely to be one of the most effective therapeutic targets.
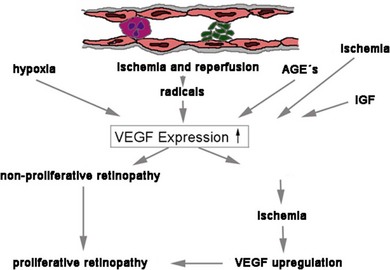
On a cellular level, VEGF has been implicated in many different mechanisms, which lead to macular edema. VEGF has, for example, been shown to decrease the proteins responsible for the tightness of the intercellular junctions and induces rapid phosphorylation of the tight junction proteins occludin and ZO-1, resulting in breakdown of the BRB.24 VEGF-induced BRB breakdown appears to be effected via nitric oxide.17 VEGF also increases paracellular transport without altering the solvent drag reflection coefficient.25 Furthermore, VEGF activation of PKC stimulates occludin phosphorylation and contributes to endothelial permeability.26
There are tight connections between inflammation and VEGF expression.17 Recently, Müller-cell-derived VEGF was shown to be essential for diabetes-induced retinal inflammation and vascular leakage.27
Investigations on cell–cell interactions by D’Amore and coworkers demonstrated an inhibitory effect of TGF-β secreted by pericytes on endothelial cell growth. In diabetic retinopathy formation of sorbitol via aldose reductase leads to PKC activation, resulting in a loss of the inhibitory balance28 (Fig. 28.8).
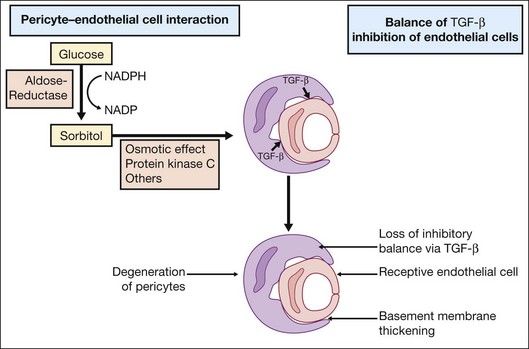
High glucose concentration leads to increased diacylglycerol (DAG) by two pathways: de novo synthesis and through dehydrogenation of phosphatidylcholine. Increased levels of DAG mediate PKC activation. Several studies have shown that a decrease in retinal blood flow occurs with PKC activation. Conversely, inhibition of PKC with LY333531 (Eli Lilly, Indianapolis, IN) normalized decreased retinal blood flow in diabetic rats.29,30
Furthermore, retinal vascular endothelial cells are very sensitive to histamine. Several studies have documented increased vascular histamine synthesis in diabetic rats and humans.31–33 The administration of histamine reduces ZO-1 protein expression and thus correlates with vascular permeability. The H1 receptor stimulates PKC that has been implicated in increased retinal vascular permeability.34 Interestingly, Aiello and coworkers showed that administration of LY333531, a PKC-β isoform-selective inhibitor, does not significantly decrease histamine-induced permeability but instead VEGF-induced permeability. In contrast, administration of nonisoform-selective PKC inhibitors did significantly suppress histamine-induced permeability.35
Furthermore, in vascular endothelial cells, advanced glycation end-products (AGE) may affect the gene expression of ET-1 and modify VEGF expression. The AGE-stimulated increased VEGF expression is dose- and time-dependent and additive to hypoxia.36,37
Endothelial cell death and vascular permeability
Where intraluminal pressure falls below a critical closing pressure the tone of the arteriolar wall cannot be maintained and the downstream capillary bed collapses and endothelial cells become “fibrin-locked.” Endothelial cells deprived of their circulation and nutrition die and only acellular basement membranes persists. A reduction in intraocular pressure may cause macular edema with cystoid degenerative changes and secondary atrophic alterations at the outer retina. Similarly, a reduction in retinal perfusion pressure, often linked to carotid/ophthalmic artery insufficiency, can have similar retinal manifestations and in extreme circumstances there may be retrograde filling of arteries from fellow veins. Stasis of the blood flow in capillaries after venous or arterial occlusions results in rapid apoptosis of endothelial cells.38
Similarly, in diabetes, retinal barrier breakdown is at least in part due to endothelial cell damage and apoptosis. The proapoptotic molecule Fas-ligand (FasL) induces apoptosis in cells that carry its receptor Fas (CD 95).39 There is evidence that FasL is expressed on vascular endothelium where it functions to inhibit leukocyte extravasation. The expression of FasL on vascular endothelial cells might thus prevent detrimental inflammation by inducing apoptosis in leukocytes as they attempt to enter the vessel. In fact, during inflammation and ensuing TNF-α release, the retinal endothelium upregulates several adhesion molecules40 that mediate the adherence of the leukocytes, but also downregulates FasL thus allowing leukocyte survival and migration to active sites of inflammation. In experimental diabetic retinopathy, inhibition of Fas-mediated apoptotic cell death reduces vascular leakage.41 The cumulative endothelial cell death during the course of diabetes plays a causal role in the pathogenesis of the diabetic vascular leakage and maculopathy.
Extracellular matrix alterations and vascular permeability
Degradation of the extracellular matrix affects endothelial cell function at many levels causing endothelial cell lability which is required for cellular invasion and proliferation, or influencing the cellular resistance and therefore the vascular permeability. The degradation and modulation of the extracellular matrix are exerted by matrix metalloproteinases (MMPs), a family of zinc-binding, calcium-dependent enzymes.42 Elevation of MMP-9 and MMP-2 expression has been shown in diabetic neovascular membranes,43,44 although a direct effect of glucose on MMP-9 expression in vascular endothelial cells could not be shown.45 It is probable that MMPs participate at various stages during the course of the BRB dysfunction and breakdown. Their actions include early changes of the endothelial cell resistance with influence on intercellular junction formation and function46 to active participation in endothelial and pericyte cell death47 that occurs late in the course of the disease.
Transcellular transport and vascular permeability
Disruption of the BRB is an early phenomenon in preclinical diabetic retinopathy. Two vascular permeability pathways may be affected, the paracellular pathway involving endothelial cell tight junctions, and the endothelial transcellular pathway mediated by endocytotic vesicles (caveolae). Despite the fact that pinocytic transport is critically involved in the transepithelial fluid exchange, its role in the pathogenesis of increased vascular leakage in diabetes is just emerging.48,49 The importance of the regulation of fluid homeostasis by active cellular transport of nutrients and fluid via pinocytosis is underlined by recent data suggesting a transient induction of the paracellular pathway and prolonged involvement of transcellular endothelial transport mechanisms in the increased permeability of retinal capillaries in diabetes.7
It is currently known that one of the factors involved in the regulation of pinocytic transport is VEGF. VEGF increases vascular permeability not only by disrupting the intercellular tight junctions between the retinal endothelial cells but also by inducing the formation of fenestrations and vesiculovacuolar organelles. The role of VEGF in the disruption of the pinocytic transport that is translated into increased vascular permeability in disease states is still controversial.50 Whereas, in higly permeable blood vessels the number of pinocytotic vesicles at the endothelial luminal membrane transporting plasma immunoglobulin G is significantly increased, no fenestrations or vesicles were found in the endothelial cells of the VEGF-affected eyes when examined by electron microscopy.
Neuronal involvement in the formation of macular edema
Recent research on DME emphasizes the role of neuronal cells in the diabetic retinal damage. The retina consists of a network of neurons and glia (astrocytes, Müller cells, and microglial cells) that comprise approximately 95% of the tissue, with blood vessels representing less than 5% of the retinal mass.51 As the network of retinal neurons and glia is intimately linked, there is no doubt that the neural (photoreceptors, bipolar cells, horizontal cells, amacrine cells, and ganglion cells) and vascular components of the retina are closely associated by metabolic synergy and paracrine communication.52,53 Neuroglial cells are involved in vision, and blood vessels provide nutrients to facilitate the process.54
In the inner retina, metabolic substrates, such as glucose, flow from vascular endothelium to astrocytes to neurons. In the outer retina, substrates reach Müller cells and photoreceptors from the choroid via the RPE.52 Microglia associate intimately with neurons that express molecules, such as CX3CL1 (fractalkine) and CD20, that negatively regulate microglial activation through their respective receptors. As such, perturbation of expression of ligand or receptor during stress would activate microglia to produce proinflammatory cytokines and acquire an activated morphology. Activated microglia produce chemokines such as monocyte chemoattractant protein-1, inducing expression of adhesion molecules, which can promote the leukostasis of neutrophils on endothelium, and potentially inducing the extravasation of inflammatory macrophages.52,54 Induction of glial fibrillary acidic protein (GFAP) is a marker of glial activation and increased expression of this protein occurs in Müller cells from the retinas of diabetic patients, but also after ischemic injury.
Mechanical factors involved in the formation of macular edema
Clinical and anatomic evidence indicates that abnormalities in the structure of the vitreoretinal interface may play an important role in the pathogenesis of DME.55–57 It was suggested that vitreoretinal adhesions in diabetic eyes are stronger than the shear forces of traction from vitreous shrinkage and this in turn may lead to the development of vitreomacular traction and subsequently to macular edema.58 Nevertheless, the risk of developing diffuse macular edema was 3.4-fold lower in the group of eyes with complete posterior vitreous attachment or complete vitreoretinal separation compared to the eyes with vitreomacular adhesion.59
The vitreous humor is a gel-like structure composed mostly of water (99%), hyaluronic acid, and collagen. A structural barrier between the vitreous cavity and the retina is formed by the inner limiting membrane (ILM), which is localized between the innermost layer of the retina and the outer boundary of the vitreous. The ILM shows typical ultrastructural characteristics of a basal lamina, is found in close contact with the foot processes of Müller cells, and contains proteins that are typically found in basal laminae such as collagen type IV and laminin.60 Striated collagen fibrils of the vitreous cortex insert into the inner portion of the ILM,61 which is also known as the hyaloid membrane of the vitreous. Detachment of the posterior hyaloid membrane with aging or pathology results in a condensation of the posterior vitreous surface (membrana hyaloidea posterior). In youth, there is adhesion between the vitreous cortex and the ILM that is stronger than Müller cells themselves and Müller cell foot processes become separated from their main cell body and remain connected to the posterior aspect of the ILM when this is separated from the retinal surface.62
There has been a controversial discussion regarding the embryonic origin of the ILM, which can be demonstrated as early as 4 weeks after gestation in the human eye.63,64 Traditionally, the ILM has been considered to be synthesized by Müller cells. This concept has been challenged by data presented from Sarthy, who investigated the expression of collagen type IV during development of the mouse eye.65 ILM proteins appear to originate largely from lens and ciliary body, although a contribution of retinal glial cells in ILM synthesis cannot be excluded. In support of this are data which show that also other ILM proteins such as perlecan, laminin-1, nidogen, and collagen XVIII are expressed predominantly in lens and ciliary body, but are not detected in the retina.66
Diffuse DME has been found in association with an attached, thickened, and taut posterior hyaloid.67 As immunocytochemical staining for cytokeratin (found in RPE) and GFAP protein (found in astrocytes and Müller cells) demonstrated the existence of cells in the premacular posterior hyaloids, suggesting a possible role for cell infiltration in the development or maintenance of macular edema. It remains to be elucidated whether these cells in the posterior vitreous cause macular edema physiologically rather than mechanically through the production of cytokines.
Treatment of macular edema
Laser treatment
Many studies have demonstrated a beneficial effect of photocoagulation therapy for DME.68–73 The exact mechanism of action of laser photocoagulation-induced resolution of DME is unknown. In short, a laser-induced destruction of oxygen-consuming photoreceptors has been discussed as well as cell death and scarring (involving gliosis and RPE hyperplasia) induced by the temporary rise in tissue temperature. Oxygen that normally diffuses from the choriocapillaris into the outer retina can now diffuse through the laser scar to the inner retina, thus relieving inner retinal hypoxia.74,75 There are contrasting data whether an increased preretinal oxygen partial pressure is involved and allows for microvascular repair in the treated areas.76,77
When studying the diameter of retinal arterioles, venules, and their macular branches before and after macular laser photocoagulation in eyes with DME, the macular arteriolar branches were found to be constricted by 20.2% and the venular branches 13.8%. This was attributed to an improved retinal oxygenation caused by the laser treatment leading to autoregulatory vasoconstriction, improving the DME.78
According to another theory, the beneficial effect of laser photocoagulation is due to an enhanced proliferation of RPE and endothelial cells leading to repair and restoration of the BRB.79 The RPE cells may respond to the injury in several ways: if the lesion is relatively small, the RPE defect can be filled by cell spreading; if the defect is relatively large, the cells can proliferate to resurface the area, and the RPE can produce cytokines (e.g., TGF-β) that antagonize the permeabilizing effects of VEGF.80,81
While focal laser coagulation reduces hypoxic areas and directly occludes leaky microaneurysms, the rationale for grid laser treatment in DME is not yet well established. Potentially, grid laser may have its beneficial effect by thinning the retina, bringing retinal vessels closer to choroidal vessels, permitting the retinal vessels to constrict by autoregulation, thereby decreasing retinal blood flow and consequently decreasing edema formation.82
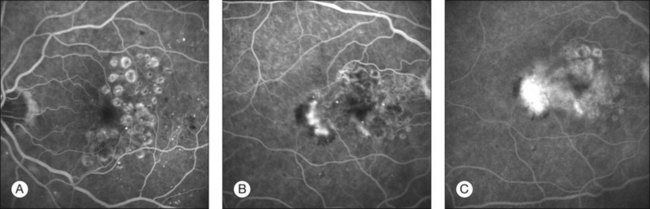
Potential side-effects are choroidal neovascularization induction (Fig. 28.9) or lack of effect in cases of diffuse edema (Fig. 28.10).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


