Chapter 42 Macular Dystrophies
 For additional online content visit http://www.expertconsult.com
For additional online content visit http://www.expertconsult.com
Introduction
The genes that cause most of the disorders in this chapter have been identified and it is tempting to try to eliminate the inconsistencies in the historical clinical nomenclature by abandoning it in favor of a scheme that is solely based on the causative genes. However, there are several serious disadvantages to such an approach. First, patients present with symptoms and signs – not with genetic test results. In addition, the near-term disease course in a given patient is usually more tightly correlated to their current clinical appearance and visual function than it is to their genotype. Thus, even in the molecular era, clinicians need to think first in terms of clinical patterns and second about the molecular mechanisms underlying these patterns. Prior to 1985 the clinical diagnosis was for all practical purposes the final diagnosis. Today the initial clinical diagnosis is really a hypothesis that has some prognostic weight by itself, and also serves to focus the search for a more precise and definitive molecular understanding of the patient’s disease through genetic testing. Clinicians are often bothered by the fact that there is an imperfect correlation between mutations in specific genes and the resulting clinical outcomes. That is, mutations in a single gene (e.g., PRPH2 and ABCA4) can cause quite different clinical appearances in different patients (e.g., retinitis pigmentosa and macular dystrophy) while a single clinical appearance (e.g., retinitis pigmentosa) can be caused by mutations in many different genes. The best way to cope with this imperfect correlation is to simply accept that the clinical diagnosis has greater validity in certain contexts while the molecular diagnosis has greater validity in others – and that the one can often strengthen the other. For example, prior to the discovery of the causative genes, characteristic clinical features were used to select genetically similar patients from heterogeneous clinic populations for the purpose of gene discovery. Now that many of the disease genes are known, genetic testing can be used to select genetically similar cohorts from clinically heterogeneous populations for the purpose of identifying the range of clinical features that can be associated with mutations in each gene. In this chapter we have taken advantage of molecular diagnosis in our patients to illustrate some unusual clinical presentations of these macular dystrophies that would have been difficult to include in such a chapter in the pre-molecular era. For the most part, we have retained the historical names associated with specific phenotypes and have grouped these whenever they are known to be caused by mutations in a single gene. We have also organized the disorders in the approximate order of their prevalence in the population with the more common diseases first. Table 42.1 summarizes the genetic characterization for all of the macular dystrophies discussed in this chapter.
The initial approach to a patient with macular dystrophy
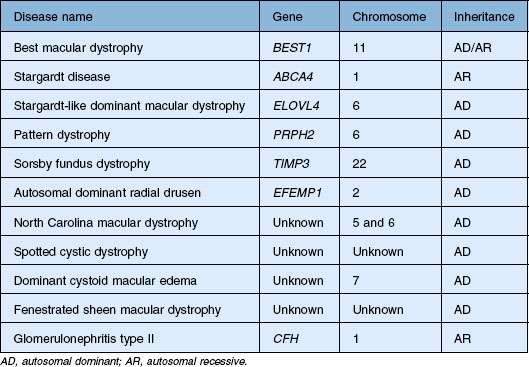
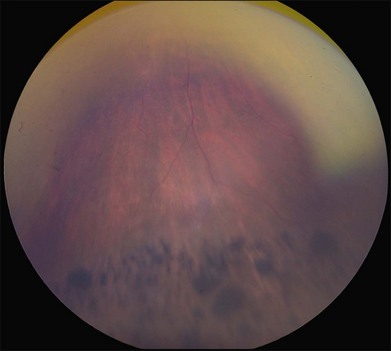
Perhaps the most important step in managing a patient with a macular dystrophy is to convince oneself that the patient truly has a Mendelian condition and not one of the many toxic, infectious, autoimmune, and multigenic disorders that can mimic them. Except in extraordinary circumstances, macular dystrophies are bilateral, and in the early years of the disease are usually extremely symmetrical in their fundus appearance. Autoimmune disorders like the presumed ocular histoplasmosis syndrome and multifocal choroiditis are often bilateral but much less symmetrical in their appearance (Fig. 42.1). Most of the macular dystrophies are inherited in an autosomal dominant fashion and thus one or more living affected relatives often exist. Identification of such a relative is one of the most powerful and most underutilized diagnostic maneuvers the clinician has at his or her disposal. One should always take a careful family history from patients suspected to have a macular dystrophy, realizing that affected individuals over the age of 50 will often carry the diagnosis of age-related macular degeneration (AMD). One should also remember that dominant macular dystrophies often exhibit variable expressivity and incomplete penetrance, such that a patient’s parents can be normal by history while more distant relatives can exhibit macular disease. Thus, when taking the history one should be very suspicious of any relative reported to have a macular disease of any kind. One should also examine any first-degree relatives who accompany the patient to the clinic and request fundus photographs and other ophthalmic records from all relatives with a history of macular disease. Not infrequently an affected relative will exhibit the “classic” features of a specific macular dystrophy but carry the diagnosis of AMD while the patient before you will exhibit a more puzzling fundus appearance and/or set of symptoms.
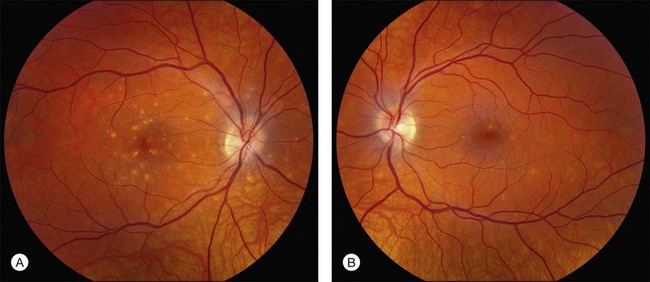
There are two classes of disease one should be especially careful to think about when initially considering a diagnosis of macular dystrophy: neuronal ceroid lipofuscinosis (NCL) and drug toxicity. In children between the ages of six and eight years, NCL (a fatal systemic disease) can present with a Stargardt-like appearance and no systemic features of any kind (Fig. 42.2). The electroretinogram is usually markedly abnormal in visually symptomatic patients with NCL but is much less likely to be abnormal in a Stargardt patient of that age. The visual dysfunction associated with NCL (both visual field and visual acuity) also tends to progress much more rapidly than Stargardt disease – over months instead of years. The other class of disease to explicitly consider and exclude in every patient suspected to have a macular dystrophy is drug toxicity. One should ask patients whether they are taking or have ever taken any medications over an extended period of time, especially medications for arthritis or skin disease (Fig. 42.3). Table 42.2 provides a list of medications that can cause macular lesions that mimic a macular dystrophy.
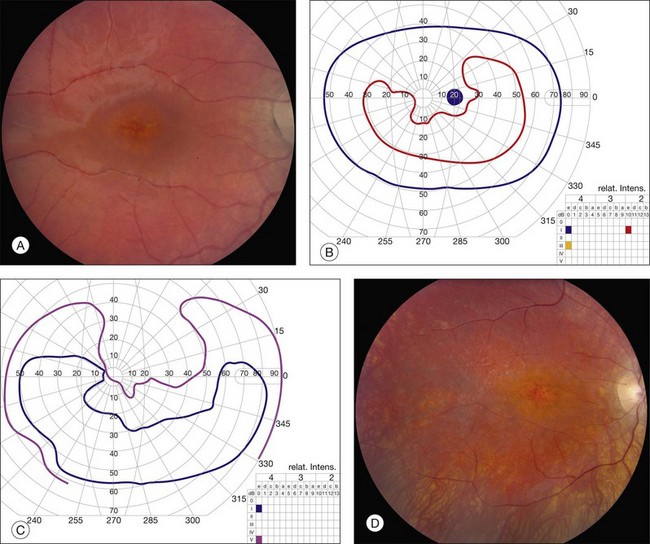
Fig. 42.3 Panels (A) and (B) depict the right eye of a 50-year-old female with plaquenil toxicity and a bull’s-eye fundus appearance that could easily be mistaken for Stargardt disease without flecks (compare with Fig. 42.30). Her visual acuity was 20/30.
Table 42.2 Drug toxicities mimicking dystrophies
| Agent | Reference(s) |
|---|---|
| Chloroquine | Hobbs et al., 1959; Marmor et al., 2011 |
| Hydroxychloroquine | Shearer et al., 1965; Marmor et al., 2011; |
| Thioridazine (mellaril) | Weekley et al., 1960 |
| Chloropromazine (thorazine) | Delong et al., 1965 |
| Clofazimine | Craythorn et al., 1986 |
| Tamoxifen | Kaiser-Kupfer and Lippman, 1978 |
| Oxalosis/methoxyfluorane | Bullock and Albert, 1975; Albert et al., 1975 |
| Canthaxanthine | Boudreault et al., 1983; Ros et al., 1985 |
| Nitrofurantoin | Ibanez et al., 1994 |
| Talc | AtLee, 1972 |
| Deferoxamine | Haimovici et al., 2002; Gonzales et al., 2004 |
Albert DM, Bullock JD, Lahav M, et al. Flecked retina secondary to oxalate crystals from methoxyflurane anesthesia: clinical and experimental studies. Trans Sect Ophthalmol Am Acad Ophthalmol Otolaryngol 1975;79(6):OP817–26.
AtLee WE, Jr. Talc and cornstarch emboli in eyes of drug abusers. JAMA 1972;219(1):49–51.
Boudreault G, Cortin P, Corriveau LA, et al. [Canthaxanthine retinopathy: 1. Clinical study in 51 consumers.] Can J Ophthalmol 1983;18(7):325–8.
Bullock JD, Albert DM. Flecked retina. Appearance secondary to oxalate crystals from methoxyflurane anesthesia. Arch Ophthalmol 1975;93(1):26–31.
Craythorn JM, Swartz M, Creel DJ. Clofazimine-induced bull’s-eye retinopathy. Retina 1986;6(1):50–2.
Delong SL, Poley BJ, McFarlane JR, Jr. Ocular changes associated with long-term chlorpromazine therapy. Arch Ophthalmol 1965;73:611–17.
Gonzales CR, Lin AP, Engstrom RE, et al. Bilateral vitelliform maculopathy and deferoxamine toxicity. Retina 2004;24(3):464–7.
Haimovici R, D’Amico DJ, Gragoudas ES, et al. The expanded clinical spectrum of deferoxamine retinopathy. Ophthalmology 2002;109(1):164–71.
Hobbs HE, Sorsby A, Freedman A. Retinopathy following chloroquine therapy. Lancet 1959;2(7101):478–80.
Ibanez HE, Williams DF, Boniuk I. Crystalline retinopathy associated with long-term nitrofurantoin therapy. Arch Ophthalmol 1994;112(3):304–5.
Kaiser-Kupfer MI, Lippman ME. Tamoxifen retinopathy. Cancer Treat Rep 1978;62(3):315–20.
Marmor MF, Kellner U, Lai TY, et al. Revised recommendations on screening for chloroquine and hydroxychloroquine retinopathy. Ophthalmology 2011;118(2):415–22.
Ros AM, Leyon H, Wennersten G. Crystalline retinopathy in patients taking an oral drug containing canthaxanthine. Photodermatol 1985;2(3):183–5.
Shearer RV, Dubois EL. Ocular changes induced by long-term hydroxychloroquine (plaquenil) therapy. Am J Ophthalmol 1967;64(2):245–52.
Weekley RD, Potts AM, Reboton J, et al.. Pigmentary retinopathy in patients receiving high doses of a new phenothiazine. Arch Ophthalmol 1960;64:65–76.
Most macular dystrophies are inherited in an autosomal dominant fashion and the identification of affected individuals in multiple generations make ABCA4-associated disease less likely. As a general rule, patients with any of the autosomal dominant macular dystrophies often have visual acuities that are better than one might expect given the striking nature of their fundus findings (e.g., Fig. 42.59) while patients with autosomal recessive Stargardt disease caused by mutations in ABCA4 often have acuities that are poorer than one might expect given the relatively mild abnormality of their fundus (e.g., Fig. 42.29).
Best macular dystrophy
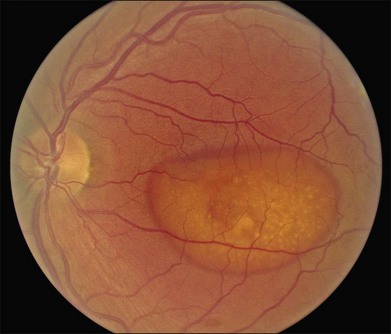
Best macular dystrophy (BMD), or Best disease, is an autosomal dominant condition caused by mutations in the BEST1 gene (OMIM #607854, formerly known as VMD2).1,2 The first family with this dystrophy was described by Friedrich Best in 1905.3 Other designations for this disease have since been used, including vitelline dystrophy,4 vitelliruptive degeneration,5 and vitelliform dystrophy.6 It is one of the most common Mendelian macular dystrophies, occurring in about 1 in 10 000 individuals. BMD refers to the “classic” form of a single, symmetric egg-yolk-like lesion centered on the fovea of each eye (Figs 42.4–42.7). However it is important to realize that BEST1 mutations are also associated with multiple other phenotypes (multifocal Best dystrophy (Fig. 42.8), autosomal dominant vitreoretinochoroidopathy (Fig. 42.9), and autosomal recessive bestrophinopathy (Fig. 42.10), which share only a few clinical features, detailed below.
Clinical features of BMD
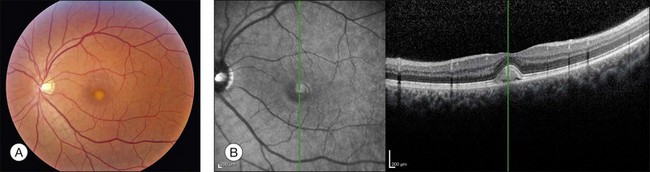
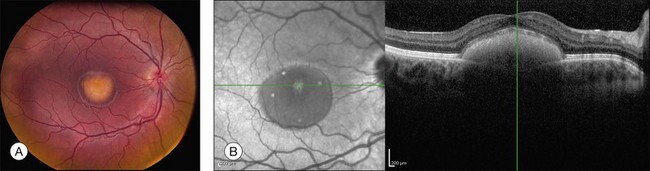
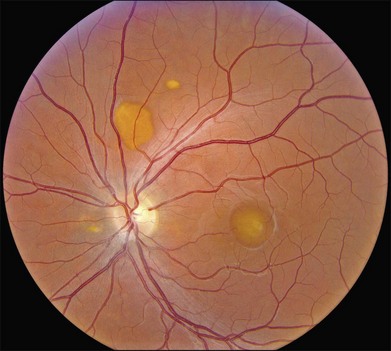
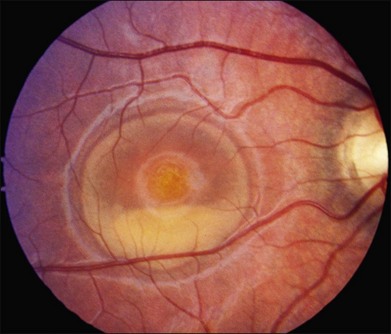
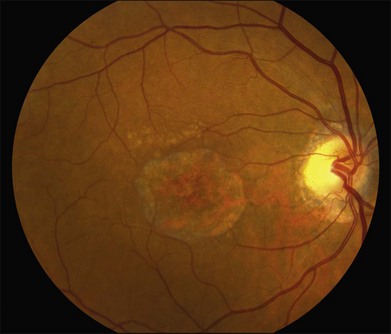
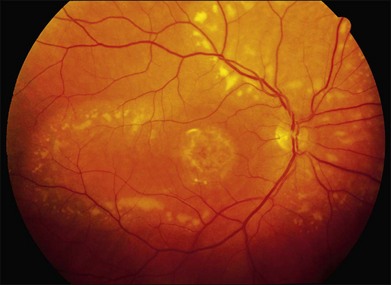
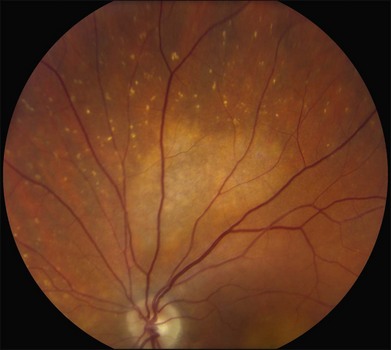
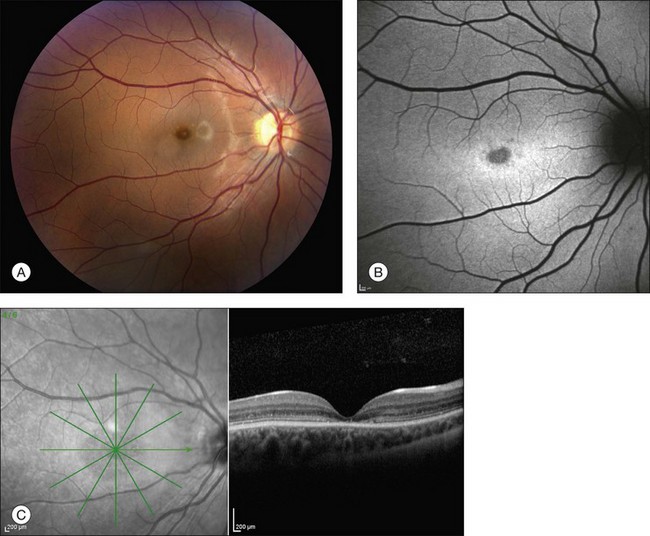
The fundus findings associated with BMD are quite varied. The macular lesions that are most characteristic of the disease are known as “vitelliform” because of their egg-yolk-like appearance (Figs 42.4–42.7). These lesions are typically solitary, round or horizontally oval, yellow, slightly elevated, and are centered on the fovea. Vitelliform lesions in patients with BMD can range in size from a few hundred microns (Fig. 42.5) to a few millimeters in diameter (Fig. 42.4). The larger lesions can be seen within the first few years of life, while the smaller lesions typically develop after age 20 and sometimes as late as age 60. As a result, the smaller lesions are sometimes referred to as “adult vitelliform” lesions (Fig. 42.5, Fig. 42.11), a phenotype that also occurs in patients with mutations in the PRPH2 gene (discussed more fully below). Some individuals who harbor disease-causing mutations in BEST1 never develop significant macular lesions.
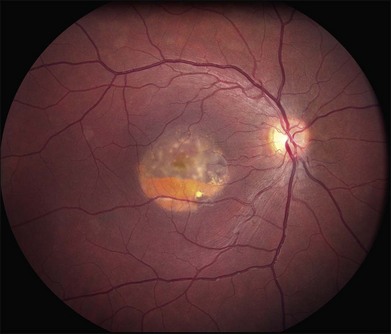
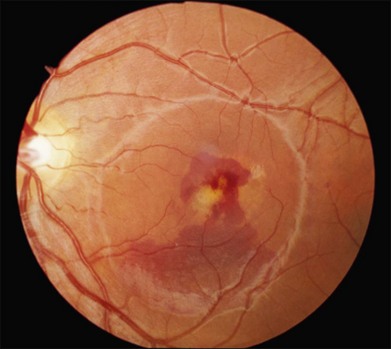
Over time, many vitelliform lesions develop a “pseudohypopyon” appearance in which the yellow material gravitates inferiorly in the subretinal space (Figs 42.12, 42.13). Other lesions develop varying amounts of subretinal and sub-RPE fibrosis, RPE atrophy in addition to hyperpigmentation, and atrophy of the RPE. This is sometimes known as a “scrambled-egg lesion” (Fig. 42.14). Many patients develop a single nodule of sub-RPE fibrosis centered very near the fovea (Fig. 42.15). Geographic atrophy is also fairly common after age 60 (Figs 42.16, 42.17), but can occur earlier in some patients. As with all diseases that disturb the RPE, true choroidal neovascularization can develop in a few percent of cases (Fig. 42.18).7 In addition, a few patients with vitelliform lesions develop subretinal hemorrhage following fairly modest blunt trauma to the head or eye (Fig. 42.19). Fortunately, these hemorrhages usually resolve and good vision returns without treatment. While these many clinical patterns have been described as “stages” by some authors, there is not always a predictable progression of these fundus changes from one to the other in a given patient. Although the maculas of patients with BMD are usually quite symmetric in the early years of the disease, the two eyes can become quite strikingly different in function and appearance as the disease progresses.
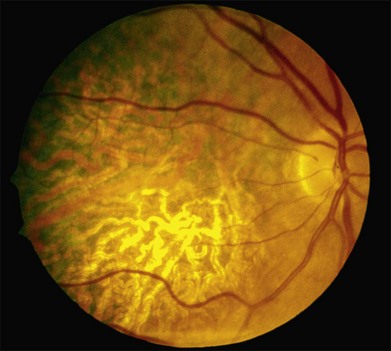
Although most patients with Best disease exhibit a single lesion centered on the fovea, there have also been numerous reports of patients with multiple vitelliform lesions who also manifest abnormalities on electro-oculography (EOG)8,9 and mutations in the BEST1 gene.10–14 In the most striking form of this multifocal phenotype, sometimes called multifocal Best dystrophy, there are multiple vitelliform lesions scattered throughout the posterior pole of both eyes (see Fig. 42.8). As with typical Best disease, these patients are asymptomatic unless the vitelliform lesions develop fibrotic scarring affecting the center of the macula. Autofluorescence and optical coherence tomography (OCT) imaging of multifocal lesions demonstrate characteristics similar to the solitary lesions seen in typical Best disease. This condition is distinguished from acute exudative polymorphous vitelliform maculopathy15,16 as patients with the latter disorder have a normal EOG and lack variations in BEST1. Notably, the canine model for BMD, which is due to a recessive mutation in the canine BEST1 gene, is associated with multifocal vitelliform lesions17,18 and some human patients with mutations in both alleles of BEST1 also exhibit multifocal vitelliform lesions.10,11,13 However, a few individuals with dominantly inherited disease and solitary lesions as children or young adults will also develop additional extramacular lesions later in life (Figs 42.20, 42.21).
Visual function
Visual acuity sufficient to drive is usually preserved in at least one eye throughout the first six decades of life, with more substantial visual loss occurring when BMD is complicated by nodular fibrosis, choroidal neovascularization19–21 or central geographic atrophy.22 Visual acuity is often 20/20 or better in eyes with undisturbed vitelliform lesions, which is surprising considering the substantial physical separation of the photoreceptor outer segments and the RPE (see Fig. 42.6) that exists for decades in some individuals (see Fig. 42.7). This suggests that the fluid within vitelliform lesions has an ionic composition relatively similar to that of the normal interphotoreceptor matrix and quite distinct from the composition of the subretinal fluid associated with rhegmatogenous retinal detachments. Some vitelliform lesions gradually flatten over time with persistence of good acuity, while others develop nodular sub-RPE scars or RPE atrophy which are associated with poorer visual acuities that are somewhat proportional to the size of the scar. Peripheral visual fields are usually completely normal in BMD, although patients with other BEST1 phenotypes do exhibit abnormal visual fields in some cases (see below).
Refractive error
It is not uncommon for patients with BMD to have hyperopia,22,23 which is likely due to shortened axial length.24 These findings are sometimes associated with narrow angles and/or angle closure glaucoma22,24,25 requiring peripheral iridotomy. Hyperopia and angle closure have also been demonstrated in other BEST1 phenotypes, including autosomal dominant vitreoretinochoroidopathy (ADVIRC)26,27 and autosomal recessive bestrophinopathy (ARB).13 Examination of the anterior segment with particular attention to the intraocular pressure and angle is warranted in patients with BMD and other phenotypes associated with BEST1 mutations.
Optical coherence tomography (OCT)
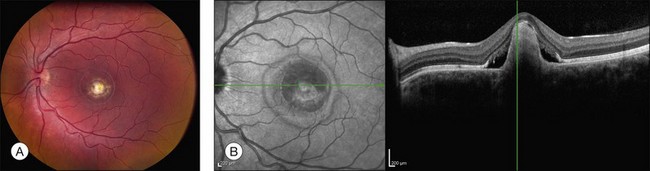
Although there have been no histopathologic studies of eyes with undisturbed vitelliform lesions, spectral domain OCT allows in vivo determination of the anatomy of macular lesions at near histopathologic resolution. The yellow material of a classic vitelliform lesion lies in the subretinal space and appears fairly homogeneous on OCT (see Figs 42.5B, 42.6B).28–32 In some patients, over time, some of the yellow pigment disappears and is replaced by clear fluid. The yellow pigment is denser than the clear fluid and settles gravitationally to the bottom of the vitelliform lesion with a fairly sharp horizontal line demarcating the pigment–fluid interface (see Figs 42.12, 42.13). This configuration is known as a “pseudohypopyon” and on OCT the yellow pigment appears hyperreflective while the clear subretinal fluid appears hyporeflective (black). Another lesion that is quite common in patients with Best disease and that has a very dramatic appearance on OCT is a fibrotic pillar that develops in the sub-RPE space, usually within 100 µm of the foveal center. These lesions appear hyperreflective on spectral domain OCT and seem to elevate the retina like a circus tent such that they are usually flanked by clear (hyporeflective on OCT) subretinal fluid (see Fig. 42.15B).
Fluorescein angiography and autofluorescence
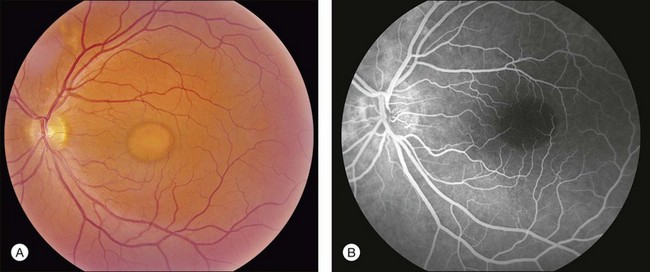
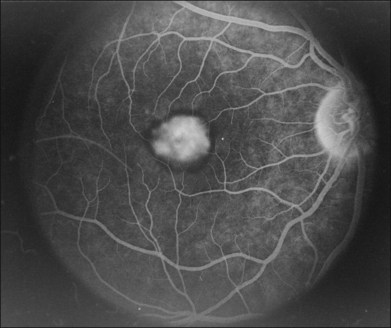
The primary clinical use of fluorescein angiography (FA) in patients with Best disease is to help differentiate non-neovascular alterations in lesion anatomy (e.g., irregular resorption of yellow pigment) from active choroidal neovascularization in patients with a recent decrease in visual acuity. Although the vitelliform lesions of Best disease resemble the pigment epithelial detachments that occur in AMD, their anatomy and composition, however, are quite different and this causes some difficulty when interpreting fluorescein angiograms in patients with Best disease. For example, the yellow pigment filling some vitelliform lesions, especially in very young patients, is extremely hydrophobic and completely excludes fluorescein from the lesion. In such patients the vitelliform lesion blocks the underlying choroidal fluorescence almost as completely as blood would in a patient with AMD (see Fig. 42.7). Over time, the contents of the subretinal vitelliform cavities in some patients become more hydrophilic and in such cases the lesions briskly and completely fill with dye during an angiogram, much as a serous pigment epithelial detachment would in a patient with AMD (Fig. 42.22). In patients with the pseudohypopyon configuration the serous component of the lesion will fill with dye while the yellow pigment in the inferior portion of the lesion will both exclude the fluorescein from the vitelliform cavity and block the underlying choroidal fluorescence. This results in an angiogram that closely resembles a “notched” pigment epithelial detachment, which in AMD patients would suggest the presence of a choroidal neovascular membrane. In patients with Best disease this appearance is much less ominous. Autofluorescence imaging is not typically needed to make the diagnosis of Best disease or to make treatment decisions. From a research perspective, however, it is interesting that undisturbed vitelliform lesions usually exhibit uniform hyperfluorescence while those that have some amount of sub-RPE fibrosis or atrophy usually exhibit hypofluorescence.22,33,34 The increased autofluorescence in the undisturbed vitelliform lesions is likely a reflection of the increased amounts of lipofuscin in eyes with Best disease.35–38
Electrophysiology
Before the discovery of the gene responsible for Best disease, electrophysiologic testing played a very prominent role in the diagnosis. The electro-oculogram is usually markedly abnormal in molecularly affected individuals, even when macular lesions are not evident. In normal individuals the cornea positive standing potential of the eye is nearly twofold higher in bright light than it is in darkness, while in patients with Best disease the ratio of the light peak to dark trough (the Arden ratio) is typically less than 1.5. Full-field electroretinography (ERG) cone and rod a- and b-wave amplitudes are usually normal. The advantage of EOG over genetic testing is that diagnostic information can be obtained within an hour or two, while the patient is in the clinic. The disadvantage is that it is usually more expensive and less sensitive than molecular testing. That is, a normal EOG does not exclude the possibility of Best disease,39–43 as 37.5% of patients in one series had a BEST1 mutation despite normal a EOG.39
Genetics
Best disease was mapped to chromosome 11q13 in 199244 and the causative gene was identified six years later.1,45,46 Now known as BEST1, this gene encodes a 585 amino acid protein known as bestrophin that localizes to the basolateral membrane of the RPE.47 To date, more than 100 different mutations in BEST1 have been associated with Best disease.1,2,22,23,34,41,42,48–62 Most disease-causing mutations are missense variants, and a substantial fraction (about 25%) occur in exon 8, suggesting that the portion of the protein encoded by this exon may be critical to its function.22 More than 90% of families that have two or more individuals with the clinical diagnosis of Best disease will have a detectable mutation in the coding sequence of BEST1. As a result, a negative molecular result is clinically meaningful and suggests that the patient’s disease is caused by another gene or that it is a non-Mendelian phenocopy. The gene that most commonly causes macular lesions that are clinically similar to those of Best disease is PRPH2 (formerly RDS), while the most common non-Mendelian phenocopy is a pigment epithelial detachment associated with age-related macular degeneration.
Pathophysiology and histopathology
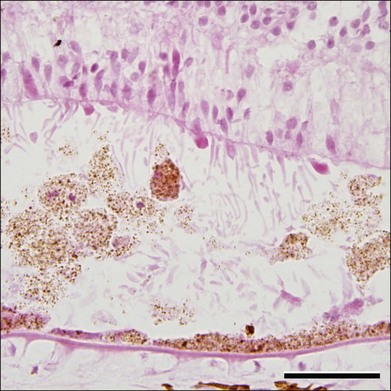
Although Best disease is rare, there have been a number of histopathologic reports describing the structural changes that accompany this condition. Histopathologic findings include increased RPE lipofuscin,37,38,63 loss of photoreceptors38 (often seen over a relatively intact RPE layer35,58), sub-RPE drusenoid material,35,36 and accumulation of cells and material in the subretinal space (Fig. 42.23). Following the discovery of the Best disease gene, efforts have also been made to identify the relationship between anatomical findings and specific genotypes. For example, eyes have been characterized from patients with Tyr227Asn,58 Thr6Arg,63 and a homozygous Trp93Cys donor.38 From these studies it has been suggested that the eyes of patients with Tyr227Asn mutations are notable for extramacular flecks.58 A donor eye with this mutation was also found to mislocalize bestrophin.58
The molecular pathophysiology of Best disease is somewhat controversial – owing in part to the difficulty of comparing the behavior of different types of cultured cells with the RPE in vivo – and a thorough discussion of the evidence for the possible functions of the bestrophin-1 protein is beyond the scope of this chapter; it has been well reviewed elsewhere.64–66 Briefly, the normal function of bestrophin-1 appears to include the regulation of the ionic milieu in the RPE and/or the subretinal space. When overexpressed in some cell types, bestrophin-1 appears to function as a calcium-sensitive chloride channel,67,68 whereas mice lacking bestrophin-169 or harboring a mutant allele (Tryp93Cys)70 show altered uptake of calcium by the RPE. It has been suggested that impaired ionic flow across the RPE could result in alterations in the adhesiveness between the interphotoreceptor matrix and the RPE or a diminution of outer segment phagocytosis, both of which are sensitive to levels of calcium.71,72
Bestrophin-1 is expressed in all RPE cells, not just those underlying the macula. Moreover, the large response of the EOG to changes in light (the Arden ratio) suggests that the entire retina participates in this response, not just the macula. Why then are the most typical Best lesions found in the macula, centered on fixation? The answer may be in part due to differences in bestrophin-1 expression in different regions of the retina.63 However, it also seems likely that there are regional differences in the adhesion of the photoreceptors to the retinal pigment epithelium. The patient shown in Fig. 42.24 is supportive of this hypothesis. She developed an aggressive epiretinal membrane that created localized traction detachments in her left eye and she developed yellow vitelliform material in each of these locations. After successful surgery to remove the membrane and relieve the traction, the extramacular vitelliform deposits persisted for years.
Additional phenotypes associated with mutations in BEST1
Autosomal dominant vitreoretinochoroidopathy (ADVIRC)
ADVIRC was first described by Kaufman et al. in 198273 as a condition with: (1) an autosomal dominant inheritance pattern; (2) peripheral pigmentary retinopathy for 360 degrees, with a discrete posterior boundary near the equator (see Fig. 42.9); (3) punctate whitish opacities in the retina; (4) vitreous cells and fibrillar condensation; (5) blood–retinal barrier breakdown; (6) retinal arteriolar narrowing and occlusion; (7) retinal neovascularization; (8) choroidal atrophy; and (9) presenile cataracts (see Fig. 42.9).74 The EOG is usually abnormal with a relatively normal ERG,75 but the first electrophysiologic studies of ADVIRC patients27,75,76 occurred in the pre-molecular era when genetic testing was not available. It has since been discovered that ADVIRC is caused by splice-altering mutations in BEST1 and that these patients can also have concomitant developmental abnormalities, including microcornea, hyperopia, and shortened axial length.26,77,78 Some patients have a severe form of ADVIRC in which both the ERG and EOG are abnormal, thus resembling retinitis pigmentosa.79,80
Autosomal recessive bestrophinopathy (ARB)
The first description of compound heterozygous BEST1 mutations causing multifocal yellowish changes in the macula and cystoid macular edema was published in 2006.11 Subsequent descriptions of patients with mutation in both BEST1 alleles have included both compound heterozygous and homozygous mutations and demonstrate a wide spectrum of fundus findings that are not present in their carrier parents.10,13,81–83 Burgess et al.13 coined the term autosomal recessive bestrophinopathy (ARB) to refer to this unusual presentation of BEST1-associated retinal disease. Hyperopia and an abnormal EOG are common. The visual acuity in ARB can be normal but tends to be worse than in autosomal dominant Best disease. Some patients exhibit cystoid macular edema or shallow subretinal fluid that can extend throughout the macula and beyond the arcades. Both solitary and mutifocal vitelliform lesions can occur, sometimes associated with flecks within and outside the macula (see Fig. 42.10). OCT can be helpful in detecting the low-lying subretinal fluid, especially in young patients who lack vitelliform lesions. Subretinal fibrosis is more common in ARB than in autosomal dominant disease.
Treatment
Treatment for BEST1 disease consists primarily of recognizing choroidal neovascularization and hastening its regression with anti-VEGF therapy. Though the natural history of subretinal hemorrhage in BMD is relatively good,19 preservation of visual function for choroidal neovascularization (CNV) has been reported in retrospective studies using intravitreal bevacizumab and ranibizumab.81,84–88 In our experience, it is not usually possible to completely eradicate subretinal fluid that exists adjacent to the nodular fibrotic pillars that occur beneath the RPE (see Fig. 42.15B). Thus, when this configuration is seen following treatment of suspected CNV, we would recommend elongating the intervals between anti-VEGF injections, and then discontinuing them altogether, once the visual acuity is stabilized and all subretinal blood has been resorbed. Even in the absence of CNV, subretinal hemorrhage can occur in patients with Best disease following relatively modest head or eye trauma (see Fig. 42.19).19,89,90 As a result, we usually caution patients against playing sports in which frequent blows to the head are to be expected. Protective eyewear is recommended for all sports. Many patients with Best disease have poor vision in one eye but retain vision sufficient in the fellow eye to drive for many years. In such individuals we recommend wearing spectacles with safety plastic lenses at all times.
Stargardt disease
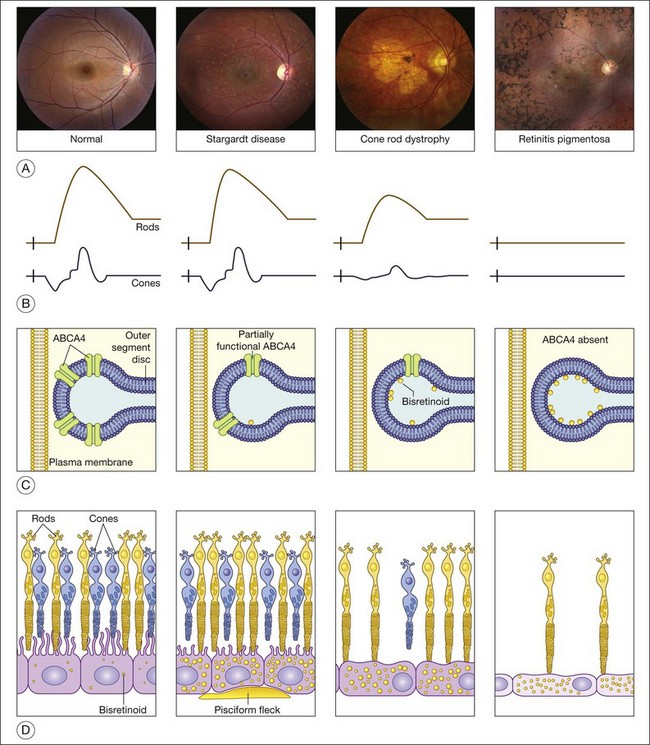
Variations in ABCA4 (OMIM #601691) are the most common cause of autosomal recessive retinal disease in humans. ABCA4 mutations were first found in patients with autosomal recessive Stargardt disease91 and were later shown to also cause cone dystrophy, cone–rod dystrophy, and retinitis pigmentosa.92–97 As discussed more fully below, a patient’s position within this disease spectrum is determined largely by the total amount of residual ABCA4 function.97 First described in 1909,98 Stargardt disease is the mildest of the ABCA4 phenotypes while a form of retinitis pigmentosa is the most severe.
Clinical features of Stargardt disease
The clinical presentation of Stargardt disease is quite variable in age of onset, presenting symptoms, and fundus appearance, and this variability is often daunting to ophthalmologists who see the condition infrequently. Most of the differences in clinical findings in patients with ABCA4 disease can be explained by the interplay of three factors that vary among patients: (1) the severity of their ABCA4 genotype (and hence the rate at which toxic bisretinoids form in the photoreceptors); (2) the relative sensitivity of the foveal cones to the genotype; and (3) the relative sensitivity of the retinal pigment epithelium to the genotype (Fig. 42.25).97 The first of these variables can be directly assessed by molecular testing of the ABCA4 gene while the molecular nature of the latter two remains to be determined.
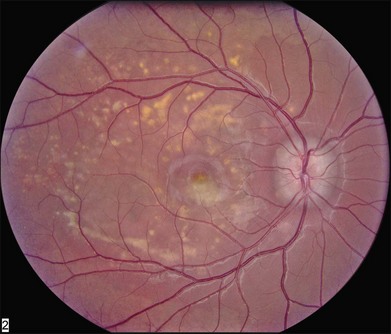
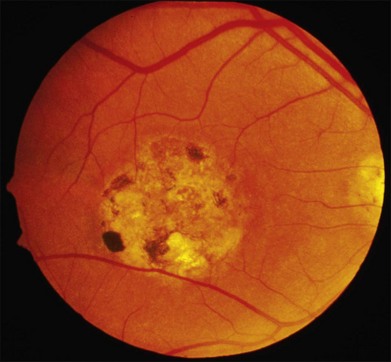
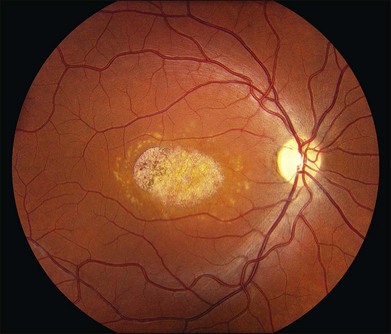
The most characteristic fundus findings in Stargardt disease are light-colored flecks at the level of the retinal pigment epithelium (Fig. 42.26). These flecks differ from drusen in that they are usually more elongated than round and they often contact each other at angles that create a branching or net-like appearance. Occasionally, two adjacent flecks form an obtuse angle that Franceschetti called “pisciform” because of its resemblance to a fish tail.99 In some patients a cluster of flecks is entirely contained within a one-by-two disc diameter horizontal ellipse centered on fixation (Fig. 42.27), while in other patients the flecks extend well beyond the temporal vascular arcades (Fig. 42.28), almost reaching the equator.
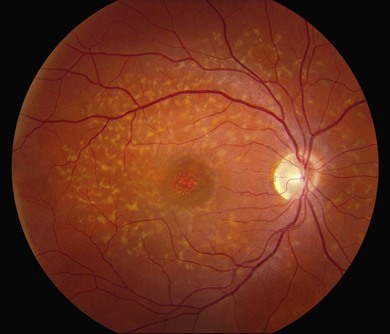
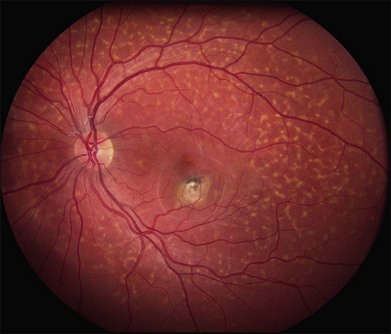
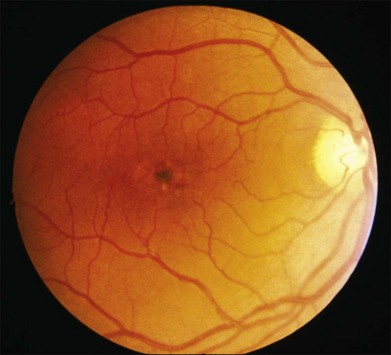
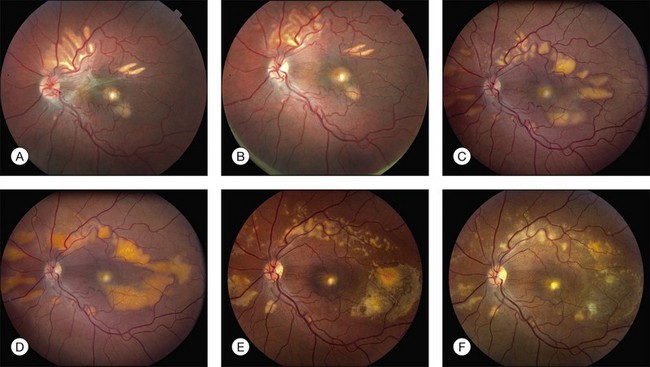
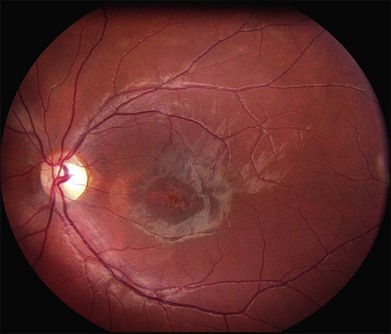
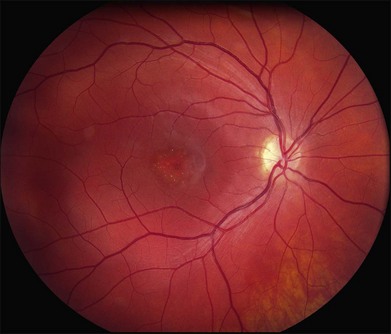
In addition to the difference in distribution, Stargardt flecks also differ widely in number, size, color, aspect ratio, and edge definition among different patients. Some patients have no flecks at all (Fig. 42.29) while others have hundreds (see Fig. 42.28). Flecks are most commonly yellow but can range from dirty-white to orange. Some flecks have pigmented edges and in a few cases this RPE hyperpigmentation can be quite dramatic. Some patients have very small deposits near the fovea that have a crystalline character on biomicroscopy (Fig. 42.30). Flecks outside the central 2 mm of the macula tend to be a bit larger than those nearer the fovea. Some patients have flecks that are almost round while others have flecks that are several times longer than they are wide. In some individuals with severe ABCA4 genotypes, the flecks are small and white and are admixed with small patches of subretinal fibrosis that resemble confetti (Fig. 42.31). This is presumably because the widespread photoreceptor injury associated with such genotypes reduces the production of bisretinoids. Some flecks are stable in position, size and number for many years, while others grow in number and/or progress to RPE atrophy (Fig. 42.32).
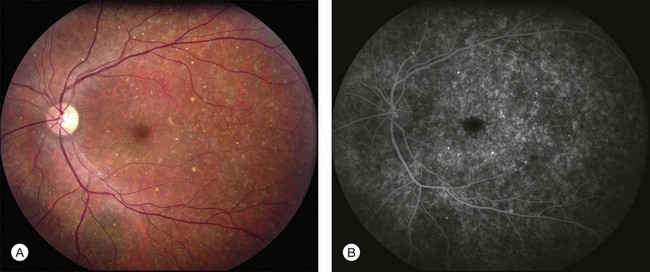
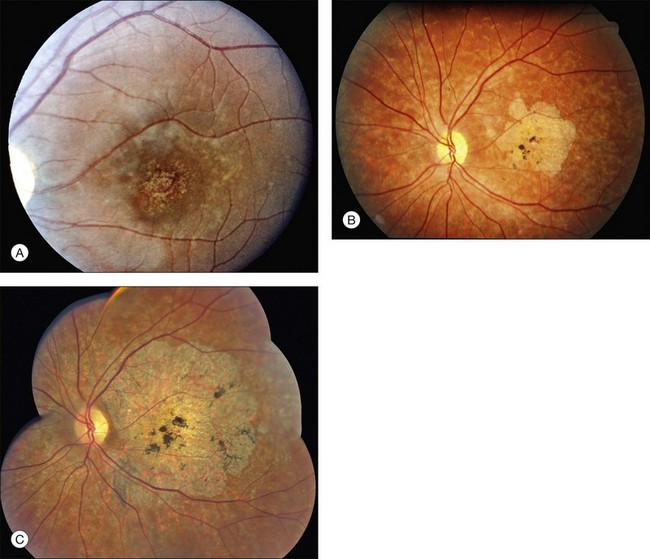
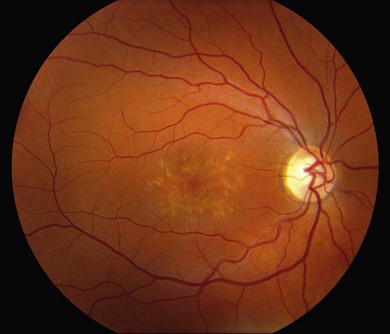
In addition to the many different fleck configurations, the RPE itself responds to ABCA4 mutations quite differently in different patients depending in part on the severity of the ABCA4 genotype and in part on the sensitivity of the RPE to the accumulation of bisretinoids. In some patients with relatively mild genotypes, there is little photoreceptor injury and thus there is a steady supply of bisretinoid to the RPE. In some patients the RPE retains this material intracellularly and as a result becomes somewhat opaque to visible light. On ophthalmoscopy this is recognized as a very uniform vermillion or light-brown color to the fundus with complete obscuration of the underlying choroidal details (see Fig. 42.27, Figs 42.33, 42.34). On fluorescein angiography, this is seen as a complete masking of the choroidal circulation. As a result, the dye-filled retinal vessels lie upon a completely hypofluorescent background (see Fig. 42.43). In other patients with a very similar bisretinoid load emanating from the photoreceptors, the RPE gradually thins, presumably from apoptotic death of some RPE cells and compensatory stretching of those that survive (Fig. 42.35). Frank RPE atrophy is commonly seen in the center of the macula and the bases of these atrophic lesions have a metallic sheen to them (Fig. 42.32) that is distinctly different from the geographic atrophy that occurs in age-related macular degeneration. In some patients these atrophic lesions become dusted with dark pigment over time (Fig. 42.35). In the later stages of disease patients with severe genotypes can develop nummular atrophy of the extramacular RPE and choriocapillaris that somewhat resembles choroideremia (Fig. 42.36).
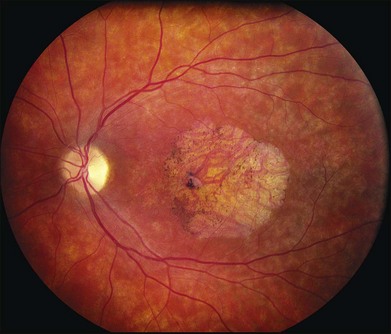
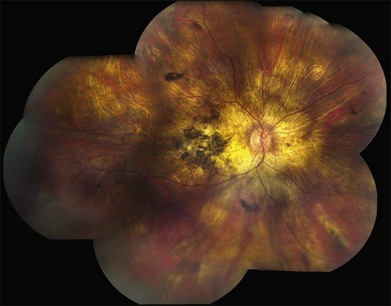
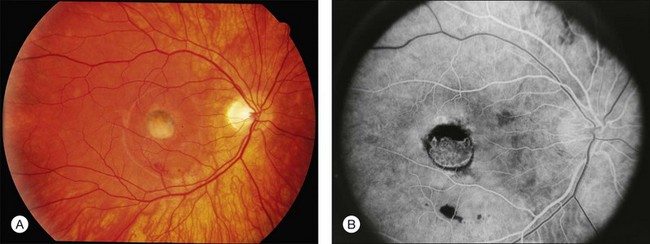
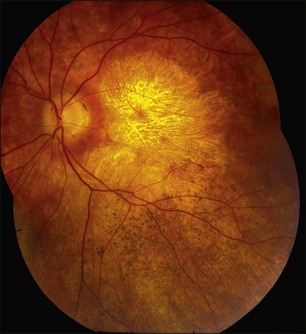
A fairly reliable diagnostic sign of ABCA4-associated retinal disease is a relative sparing of the peripapillary RPE. This sign is often more visible on fluorescein angiography100 (see Fig. 42.31) or autofluorescence;101,102 however, it can also be easily observed on ophthalmoscopy alone (Fig. 42.37). The mechanism of this sparing is currently unknown. Patients with ABCA4-associated retinal disease are not immune to inflammatory or traumatic retinal injuries and the coexistence of these disorders can make it difficult to establish the correct diagnoses. Although the frequency with which epiretinal membranes (Fig. 42.38A-D), subretinal fibrosis (Figs 42.39, 42.40) and inflammatory nodules (see Fig. 42.38A) occur in Stargardt patients may not be greater than would be expected by chance, the exuberant nature of some of these lesions suggests an adjuvant effect of either the bisretinoids themselves or the low-grade inflammation that is commonly present in degenerative retinal disease.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


