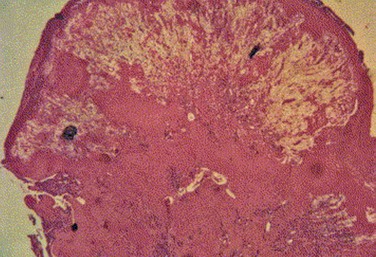
24 The first description of ligneous conjunctivitis appeared in 1847, with Bouisson1 describing a 46-year-old man with bilateral pseudomembranous conjunctivitis. In 1933, the term ‘ligneous,’ meaning ‘woody,’ was introduced by Borel2 to describe the characteristic wood-like consistency of the pseudomembranes. However, the link between ligneous conjunctivitis and plasminogen deficiency was not established until 1997 by Mingers et al.3 As many patients develop similar pseudomembranes in other mucous membranes of the body, Mingers et al.3 suggested the term ‘pseudomembranous disease’ to describe the systemic nature of this disease. Although the lesions have been referred to as pseudomembranous in the literature, the lesions are actually true membranes, as they bleed upon removal. It is characterized by multiple recurrences after local excision, and management involves the adjunctive use of topical and systemic replacement of plasminogen. Ligneous conjunctivitis is rare, and the prevalence of this disorder has not been firmly established. Ligneous conjunctivitis, associated with plasminogen deficiency is typically an autosomal recessive disorder that results from a homozygous or compound heterozygous defect. A study of 9611 blood donors in Scotland revealed a prevalence of 2.9 per 1000 heterozygous type I plasminogen deficiency subjects.4 The theoretical prevalence of homozygotes or compound heterozygotes has been calculated in the range of 1.6 per 1 million people.5 Females appear to be affected more than males, with a ratio of 1.27 : 1 to 1.39 : 1.5,6 The largest study of patients with severe type I plasminogen deficiency included 50 patients.6 The median age of clinical presentation was 9.75 months (range, 3 days to 61 years), demonstrating that ligneous conjunctivitis can occur in older individuals, despite it being classically associated with infants and children. The most common manifestation in this group was ligneous conjunctivitis (80%), followed by ligneous gingivitis (34%), with 14% of patients suffering from both. Less common extraocular manifestations included involvement of the ears, upper and lower respiratory tract (sinus, larynx, bronchi, lungs; 30%), the female genital tract (8%), the gastrointestinal tract (duodenal ulcer; 2%), congenital occlusive hydrocephalus (8%), and juvenile colloid milium of the skin (2%). Two of the patients with congenital occlusive hydrocephalus had Dandy–Walker malformation. Several studies have shown extraocular manifestations of severe plasminogen deficiency without ocular involvement.5,6 There has been definitive evidence to support that ligneous conjunctivitis is the result of plasminogen deficiency.7–10 The case of ligneous conjunctivitis reported by Mingers et al. was also the first report of plasminogen deficiency in humans.3 There are two types of plasminogen deficiency: type I (hypoplasminogenemia) is a quantitative deficiency and type II (dysplasminogenemia) is a qualitative deficiency. Type I deficiency is the type most associated with ligneous conjunctivitis. Development of ligneous lesions is most commonly caused by sporadic mutations in the plasminogen gene; however, compound-heterozygous or homozygous mutations have been reported.3,5,6,9,10 Tefs et al. found the K19E mutation to be the most common genetic cause of type I plasminogen deficiency (34%) in a series of 50 patients.6 Knowledge of the genetics of the disease allows for prenatal diagnosis in known carrier families, which can be crucial in cases of obstructive congenital hydrocephalus.11 Plasmin is a serine protease and is the predominant fibrinolytic enzyme in the human circulation, it is also found in the extracellular matrix.12 Plasminogen is converted to plasmin by plasminogen activators in the blood. Plasmin plays an important role in hemostasis as well as being an integral component of wound healing in its role in degrading fibrin. The gene for plasminogen is located on chromosome 6, and is produced predominantly by the liver. With plasminogen deficiency, wound-healing capability is diminished and is most pronounced in mucous membranes, such as the conjunctiva.12 The impaired wound-healing capacity causes an arrest at the stage of granulation tissue formation and excessive fibrin deposition. Thus, fibrin-rich membranes or mucus strands accumulate, stimulating inflammatory cells and fibroblasts, while desiccation of the fibrin leads to the ligneous consistency of the conjunctival lesions. Similar pathophysiologic mechanisms occur in extraocular manifestations of plasminogen deficiency. While in extravascular spaces, fibrinolysis with low to nonexistent levels of plasminogen activity is impaired. However, intravascularly this is not the case, as can be inferred from the fact that thrombotic phenomena are absent in patients with ligneous conjunctivitis and plasminogen deficiency. It has been suggested that nonplasmin-induced fibrinolysis is intensified in patients with plasminogen deficiency and ligneous conjunctivitis, through elevated polymorphonuclear elastase levels, among other factors.10 Due to the appearance, histopathology, clinical course, and response to treatment, the authors also believe that ligneous conjunctivitis results from an exaggerated inflammatory response to tissue injury. This injury may arise from infection or physical trauma, including surgery. These factors may incite a genetic predisposition, such as plasminogen deficiency to develop this response. Schuster and Seregard also postulated that conjunctivitis is the most common manifestation of plasminogen deficiency, due to frequent exposure to ocular irritants.5 These irritants may start or perpetuate local inflammation and create ligneous membranes. Many cases of antecedent viral or bacterial infections have been described in the literature, including staphylococcal, streptococcal, and Haemophilus conjunctivitis.13–15 In these cases, it appears that ligneous conjunctivitis in genetically susceptible individuals develops as an abnormal response to the conjunctival trauma elicited by the infecting organisms. Trauma, especially from surgery, is also thought to be a cause of ligneous conjunctivitis. The authors have reported a case of a 24-year-old woman with ligneous conjunctivitis of the left upper eyelid, who underwent a conjunctival autograft from her left lower to left upper eyelid. Ligneous conjunctivitis subsequently developed at the previously unaffected donor site and became resistant to treatment in the original site of disease in the palpebral conjunctiva of the upper lid.16 Later studies reported the same negative experience in previously unaffected fellow eyes.17 In these cases, ligneous conjunctivitis appears to develop as an abnormal response of the immune system to the conjunctival trauma. Histologic examination of ligneous membranes shows superficial or subepithelial deposits of eosinophilic amorphous hyaline, amyloid-like material with a variable proportion of granulation tissue with accompanying inflammatory cells (lymphocytes, plasma cells, and granulocytes) (Fig. 24.1).5 This amorphous hyaline-like material has been shown to contain mainly fibrin and other plasma proteins, such as albumin and immunoglobulins (mainly IgG).5 Ligneous lesions may also contain variable amounts of mucopolysaccharides in adjacent granulation tissue.5 Abnormal vascular permeability has also been suggested as the source of the various components of the ligneous lesion. Melikian postulated that a serofibrinous transudate from the conjunctival neovascularization undergoes subsequent coagulation, with the resulting formation of granulation tissue and accumulation of the hyaline material, which becomes hardened in forming the ligneous membranes.18 Immunohistochemical evaluation of ligneous conjunctivitis lesions was first performed in 1988.19 In this study, a cellular infiltrate was seen that was composed mainly of T lymphocytes, which was seen again in later studies.17 The ratio of T-helper/inducer : T-suppressor/cytotoxic cells was approximately 3 : 1. Immunofluorescent techniques demonstrated that the major components of the hyaline material in the substantia propria were immunoglobulins. IgG was the most prominent, with staining for both light and heavy chains, but primarily for κ-light chains. Topical cyclosporine A has been reported to be more effective than other topical agents (short of plasminogen preparations) in the treatment of this disorder in several studies.15,16,19 Cyclosporine A interferes with IL-2 production, therefore, preventing the activation and recruitment of the T-cell response. Immunohistochemical analysis of a lesion resected after 6 months of topical cyclosporine A therapy, supports the immune role of this entity.19
Ligneous Conjunctivitis
Introduction
Epidemiology
Etiology
Pathophysiology/Histopathology
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Ento Key
Fastest Otolaryngology & Ophthalmology Insight Engine