Chapter 88 Latent nystagmus and dissociated vertical–horizontal deviation
LATENT NYSTAGMUS DISTINGUISHING CHARACTERISTICS AND ASSOCIATIONS
BINOCULAR DECORRELATION FROM VARIOUS CAUSES BEGINS THE LN CASCADE
MALDEVELOPMENT IN V1 IS PASSED ON TO AREAS MT/MST
BINOCULAR DECORRELATION UNMASKS AN INNATE NASALWARD MONOCULAR BIAS
HYPOTHETICAL SIGNAL FLOW FOR LN
MECHANISM OF DISSOCIATED VERTICAL DEVIATION: DAMPING VERTICAL AND CYCLOTORSIONAL COMPONENTS OF LN/MLN
MECHANISM OF DISSOCIATED HORIZONTAL DEVIATION: EXODEVIATION WHEN CONVERGENCE DAMPING OF LN/MLN RELAXES
HOW HEAD POSTURES AFFECT LM/MLN AND DVD
Latent nystagmus distinguishing characteristics and associations
Latent nystagmus (LN) is a common subtype of pathologic nystagmus observed in humans and NHPs.1 It is linked strongly to binocular maldevelopment in infancy, either from strabismus or amblyopia.
LN is characterized by a conjugate, horizontal slow-phase drift of eye position that is directed nasalward with respect to the viewing eye.2,3 When viewing switches from eye to eye, the direction of the slow-phases reverse instantaneously: leftward when the right eye is fixating and rightward when the left eye is fixating (Fig. 88.1). The severity of the nystagmus (and its visibility during clinical examination) increases when one eye is covered, hence the term “latent.” When the nystagmus is evident with both eyes open, it is called manifest LN (MLN).
LN is distinguished from congenital (“idiopathic infantile”) nystagmus (CN, or the infantile nystagmus syndrome – INS) by the feature of instantaneous reversal-of-direction with alternating fixation. By eye movement recording, it is distinguished also in waveform which is always that of decreasing velocity and linear trajectory, whereas that of CN/INS is of increasing velocity and pendular trajectory.2 Eye movement recordings, or high magnification clinical inspection with a slit-lamp or ophthalmoscope, frequently reveals a superimposed small torsional movement.
Our understanding of the clinical features of LN have been clarified by Dell’Osso et al.,2,4 who have described the historical origins of LN’s various terms. In 1872, Faucon5 first described manifest LN (MLN). In 1912, Fromaget and Fromaget6 introduced the term “nystagmus latent.” These early reports of LN were reviewed by Sorsby7 in 1931. The oxymoron “manifested latent” nystagmus was introduced by Kestenbaum8 in 1947, who emphasized that LN is often observed in patients with strabismus when they view with both eyes open.
Although infantile esotropia is the leading association with LN, any disorder that perturbs development of binocular fusion in infancy, such as monocular or severe binocular deprivation, will produce LN and MLN.9,10 The NIH Committee on Eye Movement and Strabismus (CEMAS) classification11 recommended that the terms LN/MLN be replaced by the etiologic descriptor fusion–maldevelopment nystagmus.
Development of fusion eliminates nasalward visual cortex biases
The postnatal development of binocular sensory and motor functions in normal infant monkeys parallels that of normal infant humans, but on a compressed time scale; 1 week of monkey development approximates 1 month of human.12–15 Binocular disparity sensitivity and binocular fusion are absent in human and monkey neonates. Stereopsis emerges abruptly in humans during the first 3–5 months of postnatal life,16–20 and in monkeys during the first 3–5 weeks,14 achieving adult-like levels of sensitivity.
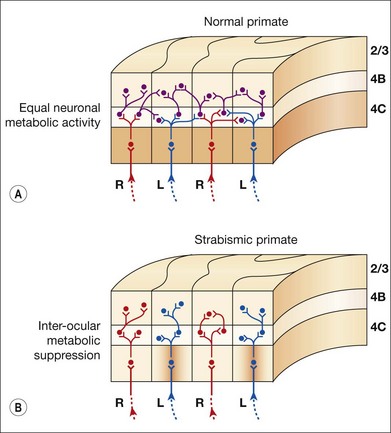
V1 horizontal axonal connections are key components of fusion development and maldevelopment (Fig. 88.2). Binocularity in primates begins with horizontal connections between V1 ocular dominance columns (ODCs) of opposite ocularity.21–23 These connections are immature in the first weeks of life, conveying crude, weak binocular responses.24–26 Maturation of binocular connections requires correlated (synchronous) activity between right eye and left eye geniculostriate inputs.27,28 Decorrelation of inputs (Fig. 88.3), produced by binocular non-correspondence, causes loss of horizontal connections over a period of days in V1 of kittens.27,29 Similar losses occur over a period of weeks in V1 of monkeys, and over a period of months in V1 of children. Binocular decorrelation also promotes interocular suppression (Fig. 88.4) as a further hindrance to fusion.1
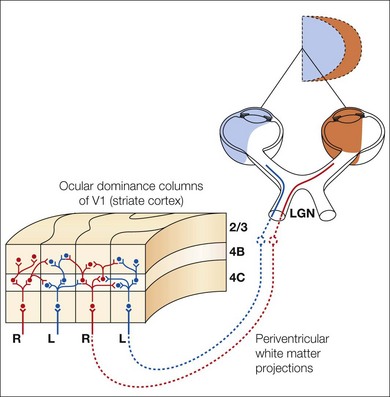
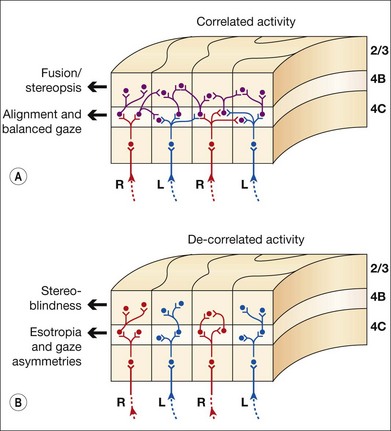
In the first months of life in humans and weeks of life in monkeys, monocular motion VEPs (visually evoked potentials) reveal a nasotemporal asymmetry.30–33 Monocular preferential-looking testing reveals greater perceptual sensitivity to nasalward motion.34 Monocular pursuit and optokinetic tracking both reveal biases favoring nasalward target motion.12,35–38 These nasalward motion biases are pronounced before onset of sensorial fusion and stereopsis, but systematically diminish thereafter. They are retained in subtle form in normal adult humans, and can be unmasked using contrived, monocular stimuli.39,40 If normal maturation of binocularity is impeded by eye misalignment or monocular deprivation, the nasalward biases persist and become pronounced.34,41–47 The nasalward gaze bias is the key feature of the fusion maldevelopment syndrome. Other common findings are loss of stereopsis, interocular suppression, strabismus, and smaller amplitude torsional/vertical oscillations of the eyes.
Binocular decorrelation from various causes begins the LN cascade
Clinical studies of children43 and adults2,4,44,48 with LN have inspired a series of behavioral, physiologic, and neuroanatomic studies in NHPs who had LN associated with naturally occurring22,23,49–55 or experimentally induced1,10,56–65 infantile strabismus. The common finding is that the prevalence and severity of LN correlates with the age of onset and duration of binocular decorrelation in infancy.
The most common clinical cause of binocular decorrelation is strabismus, which in human infants is overwhelmingly esotropic (convergent).66 Early-onset esotropia exceeds exotropia by a ratio of 9:1. Esotropia is also the most common form of naturally occurring strabismus in NHPs.67,68 However, any prolonged deprivation of normal binocular experience in early infancy (monocular congenital cataract, uniocular high hyperopia, myopia or astigmatism, uniocular neonatal vitreous hemorrhage, uniocular corneal clouding, dense bilateral cataracts, NHP models) can cause binocular decorrelation, including monocular deprivation (uniocular amblyopia) or severe binocular deprivation (bilateral amblyopia),10,57,63 eyelid suturing (the thin, translucent eyelid of NHPs mimics a congenital cataract, allowing diffuse luminance to the retina but blocking spatial vision). Loss of spatial vision is not required; the majority of human and NHP infants with strabismus alternate fixation initially and have no amblyopia.69 The necessary and sufficient factor is binocular decorrelation, not lack of sharp visual acuity.
Decorrelation durations that exceed the equivalent of 3 months in human infant development result in an LN prevalence of 100%.1,65,70 Perturbing these inputs from week 1 of life causes LN, but delaying the perturbation to the time of onset of normal fusion and stereopsis (the equivalent of age 2–4 months in humans) is equally effective.71 The severity of the resultant LN corresponds to the severity of loss of binocular connections between ODCs of opposite ocularity in visual area V1, and the severity of interocular suppression.1,72 Area V1 feeds forward to extrastriate areas (areas MT/MST) important for gaze-holding and gaze-tracking, such as smooth pursuit, OKN, and the short-latency ocular following response.73–76
Maldevelopment in V1 is passed on to areas MT/MST
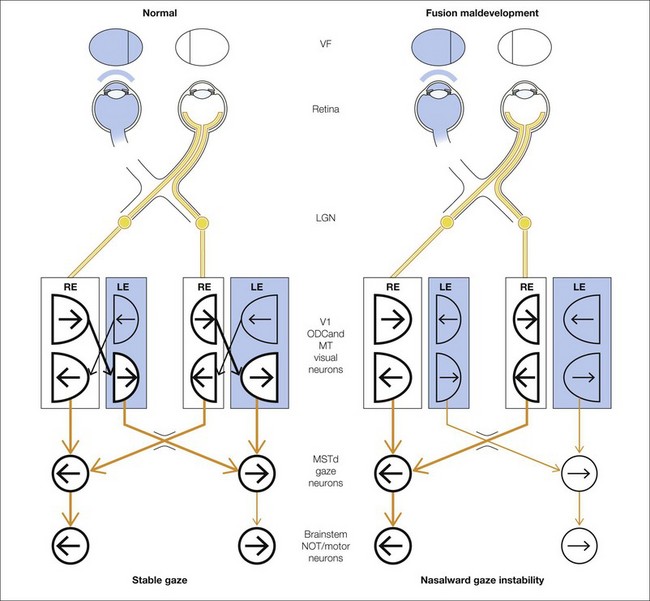
Visual areas V1, V2 (prestriate cortex), MT (medial temporal), and MST (medial superior temporal) of the cerebral cortex are major components of the conjugate gaze pathway.77 Each of these areas in normal primates contains directionally selective, binocular neurons.78–81 MST in each cerebral hemisphere encodes ipsiversive gaze.74,82–84 MST projects downstream to the brainstem visuomotor nuclei that generate eye movements: the NOT, medial vestibular nucleus, and interconnected abducens and ocular motor nuclei.77,85 In primates, subcortical inputs to NOT may play a minor role.9,10,86 The dominant pathway is cerebral – from MST to brainstem. The dominant role of the cortical pathway, and the minimal role of a subcortical pathway, is reinforced by studies of children. Neuroimaging of visual cortex, combined with eye movement recordings, have shown absence of visually driven pursuit or OKN in cerebrally blind infants.87,88
One mechanism for the gaze-holding asymmetry would be over-representation of nasalward neurons within visual areas V1 through MT in the immature/strabismic cortex. However, directional and binocular responses of neurons in V1, V2, and MT have been investigated in infant monkeys, as well as in monkeys with early-onset strabismus, and no over-representation of neurons selective for nasalward motion has been found.26,61,89,90 In strabismic animals, binocular (excitatory) responses are reduced and interocular suppression is increased.89–91 These physiologic abnormalities have neuro-anatomic correlates. In V1 of strabismic monkeys, binocular connections are deficient22,23 and interocular metabolic activity is suppressed.53,92,93
Binocular decorrelation unmasks an innate nasalward monocular bias
LN is always linked to abnormal binocular development in infancy. This important clinical observation motivated the studies of NHPs reviewed above, which have in turn provided the functional-structural correlations needed to explain the pathophysiology of LN. The NHP studies have motivated pediatric ophthalmologists to repair fusion earlier in infancy,94 thereby preventing LN or reducing its severity.
Latent nystagmus is caused by an afferent binocular visual pathway defect. The binocular defect unmasks a directional bias encoded in the cerebral gaze pathways. Normal binocular development (fusion) – in the first months of life – eliminates the directional bias; abnormal development (maldeveloped fusion) exaggerates the bias. If fusion goes unrepaired in infancy, the directional bias persists throughout adult life.1,95
The visual cortex in each cerebral hemisphere is wired innately for nasalward motion. The innate wiring is monocular. To generate temporalward gaze-holding, signals must traverse binocular connections, unimpeded by interocular suppression. If normal binocularity fails to develop, the system remains predominantly monocular and asymmetric, incapable of driving temporalward gaze-holding or robust, temporalward pursuit/OKN.10,43,44,61,66,90 LN is an abnormal monocular bias added on to a normal, ipsiversive hemispheric gaze bias.
Hypothetical signal flow for LN
Figure 88.5 illustrates the mechanism for LN, showing the circuit mediating gaze-holding in primates and the role of binocular connections. Shaded structures indicate less active visual and motor neurons caused by occlusion of one eye and/or interocular suppression. The circuit on the left depicts the pathways and visuomotor component structures in a primate with LN.