Ischemic Stroke
Jose G. Romano
EPIDEMIOLOGY
In spite of decreasing rates of stroke mortality in the last decades, with 700,000 new strokes each year and 160,000 annual deaths, stroke is the third cause of death and leading cause of disability in the United States.1 The cost of stroke to the health care system is over $40 billion,2 but the cost in human suffering is difficult to estimate. Seventy percent of the estimated 4 million stroke survivors are vocationally disabled,1 approximately 30% fulfill criteria for dementia,3,4 and a similar number have major depression.5,6 The elderly are particularly vulnerable, because age is the most important risk factor for stroke. Stroke incidence triples with each decade after age 35, and seven of eight stroke deaths occur in those age 65 years or older. The magnitude of the problem can be put in perspective by the estimation that the population older than 65 years will increase by 50% from 1995 to 2025.7 Stroke fatality varies according to stroke subtype, but on average 20% to 25% of those with ischemic strokes and 50% of those with hemorrhagic strokes will die within 30 days of the event.8 However, new understandings of stroke mechanisms and acute revascularization techniques have created optimism to counter these ominous figures.
Stroke is not one disease but a group of conditions that result in sudden brain injury caused by alterations of cerebral blood flow. This review focuses on ischemic stroke, which accounts for 80% of all strokes, and emphasizes those areas of particular interest to ophthalmologists.
STROKE SIGNS AND SYMPTOMS
LARGE VESSEL MANIFESTATIONS
The carotid arteries carry 80% of cerebral blood flow, irrigate the territories of the middle cerebral, anterior cerebral, and anterior choroidal arteries, and provide blood flow to the eye through the ophthalmic artery. The symptoms of carotid embolism vary according to the recipient artery. Occlusion of the ophthalmic artery or its branches results in monocular visual loss (amaurosis fugax, central or branch retinal artery occlusion), often described as blurring, graying or curtaining of vision.9 In addition, in those with chronic flow deprivation to the retinal circulation, transient and brief monocular visual loss in response to bright light can be seen.10 Middle cerebral artery syndrome often includes contralateral weakness affecting the face and arm more than the leg, contralateral sensory loss, and aphasia if the dominant hemisphere is affected, or hemi-inattention/neglect with nondominant lesions. In addition, there may be gaze deviation toward the affected hemisphere.11 Anterior cerebral artery strokes affect the contralateral leg and shoulder more than face and arm, and can have associated abulia and incontinence.12 The anterior choroidal stroke affects the basal ganglia and internal capsule, with contralateral hemiplegia, hemisensory loss, and hourglass-shaped homonymous visual field defects.13 Anterior choroidal infarcts may be difficult to differentiate from lacunar strokes because of occlusion of small penetrators.
Clues for carotid disease on examination include an audible bruit in the anterior neck, evidence of a cholesterol retinal embolus (Hollenhorst plaque), and a history of various spells of ischemia in the same distribution. In addition, careful craniovascular examination may reveal asymmetric preauricular and orbital pulses, which are stronger on the side of the stenosis, representing external carotid artery collateral flow. On occasions, dilatation of the extracranial vasculature may be noted over the forehead.
The posterior circulation accounts for 20% of embolic strokes. Vertebral syndromes may affect the medulla or cerebellum as these structures are fed by branches of the vertebral arteries, or may cause distal embolization into the basilar and posterior cerebral arteries.14 The most common medullary manifestation is the posterolateral medullary syndrome or Wallenberg syndrome, caused by occlusion of the vertebral artery or the posterior inferior cerebellar artery (PICA). It is characterized by vertigo, nausea, vomiting, miosis and ptosis, ipsilateral face and contralateral hemibody numbness, hoarseness, and dysphagia; hiccups may also be present. When the PICA is involved, there are also cerebellar manifestations, with consequent hemidysmetria and gait ataxia. The medial medullary syndrome caused by distal vertebral occlusion, results in hemiparesis and tongue deviation. When the basilar artery occludes, there is usually the combination of extraocular movement abnormalities, descending motor fiber involvement with hemi or quadriparesis, and alteration of the state of consciousness. A comatose state is common in basilar artery occlusion, but large hemispheric strokes may impair consciousness through increased intracranial pressure, as do large cerebellar strokes that press on the brainstem and result in hydrocephalus. Occlusion of the posterior cerebral artery causes visual field loss by virtue of occipital injury. The proximal posterior cerebral artery also feeds the thalamus and the medial temporal lobes, and therefore there may also be sensory, memory, and behavioral manifestations.15
On general physical examination it is important to record the blood pressure in each arm and compare both radial pulses. Differential blood pressure or radial pulse, as well as a bruit in the supraclavicular area, constitute indirect evidence of subclavian and possibly vertebral origin disease.
Border zone infarcts resulting from hemodynamic failure as a consequence of large vessel flow obstruction may affect the anterior frontal as well as the corona radiata in a longitudinal fashion (border zones between the anterior and middle cerebral arteries) or the parietooccipital area (border zone between the middle and posterior cerebral arteries).16 The classic description of bilateral border zone infarcts that may be seen after significant hypotension is the “man in the barrel” syndrome, where proximal upper and lower limb weakness is noted.17 More commonly, the border zone is unilateral and the manifestations are often similar to those seen in embolic strokes.18
Headache as a symptom of cerebral ischemia occurs as an initial manifestation of carotid or vertebral artery dissection, on occasions accompanies the acute occlusion of a large artery such as the vertebral and basilar artery, is common in stroke caused by giant cell arteritis, and may develop later in the course of ischemia as a result of increased intracranial pressure.19
SMALL VESSEL MANIFESTATIONS
The manifestations of small penetrating artery occlusion depend on the location of injury. The lacunar syndromes usually result from single small perforating artery damage, but larger strokes, such as a striatocapsular stroke caused by proximal middle cerebral artery occlusion with infarct in the distribution of multiple penetrating arteries, may have similar clinical findings. The classic lacunar syndromes include pure hemiparesis (internal capsule), pure hemihypoesthesia (thalamus), hemiparesis with hemihypoesthesia (thalamocapsular), ataxic hemiparesis (corona radiata and pons), dysarthria with a clumsy hand (corona radiata and pons), but many other less common syndromes have been described.20 When small basilar penetrator arteries occlude, there may be diplopia, hemiparesis, perioral numbness, ataxia, and dysarthria. The accumulation of multiple small subcortical infarcts results in a range of cognitive impairments, including dementia.21 Dementia from cerebrovascular disease may also be caused by single strategic strokes (such as caudate or thalamic strokes) or multiple cortical strokes.22 In addition, right parietal strokes and dominant hemisphere temporal strokes (with fluent aphasia) may be confused with psychiatric conditions.
VENOUS STROKES
Thrombosis of cerebral veins, or the larger cerebral sinuses, causes symptoms both by its location and by increased intracranial pressure. These infarcts commonly have a hemorrhagic component because venous outflow obstruction results in extravasation from the capillary bed. Blood products are quite irritative and therefore seizures frequently accompany venous infarcts. When large cerebral sinuses are occluded, particularly the superior sagittal sinus, increased intracranial pressure may result. This syndrome is characterized by headaches, papilledema, and occasionally abduscens nerve palsy, and may be confused with benign intracranial hypertension or pseudotumor cerebri. When the cavernous sinus is affected, ipsilateral proptosis, oculomotor palsy and trigeminal involvement (of its V1 and sometimes V2 segments) is noted.23
TRANSIENT ISCHEMIC ATTACKS
An episode of sudden neurologic dysfunction because of cerebral ischemia that is transient and leaves no observable sequelae is denominated a transient ischemic attack (TIA). Although the duration of such spells has traditionally been defined as less than 24 hours, this concept is currently being reconsidered to events of 1 hour or less.24 The average duration of a TIA is 14 minutes for the anterior circulation and 8 minutes for the posterior circulation.25 In those whose symptoms last 1 hour or more, only 14% have complete recovery by 24 hours,26 and magnetic resonance imaging (MRI) studies have revealed small strokes in half of those with transient neurologic symptoms.27
When cerebral blood flow decreases below approximately 20 mL per 100 g of brain tissue per minute, neuronal function will cease and symptoms will become manifest.28 If blood flow is restored promptly, usually because of the body’s endogenous thrombolytic ability, then full function will be restored. Therefore, transience of symptoms depends in large part on the size and composition of the responsible embolic particles, as well as the robustness of collateral flow.
Cardioembolic strokes commonly debut with strokes, as the emboli produced in the cardiac chambers are usually large: the diameter of the middle cerebral, vertebrals, and basilar arteries is approximately 3 mm, and it is not unusual for cardiac emboli to be larger than this. In comparison, the platelet-rich emboli formed in high-shear rate areas such as stenotic arteries are much smaller and therefore TIA is a frequent predecessor to aortic, carotid, and vertebral strokes. This phenomenon of continuous or repetitive embolization is illustrated by monitoring the cerebral arteries with transcranial Doppler ultrasonography, where the detection of small microembolic signals distal to carotid stenosis is common.29,30 Presumably, many of these signals represent small emboli that are washed into the venous circulation,31 but their presence predicts plaque instability and risk of stroke. Embolism is not the only cause of TIA, which may occur in the territory distal to a fixed arterial stenosis or occlusion in the setting of hypotension or hypovolemia, with resolution of symptoms with increased flow. Recurrent TIAs with similar manifestations are suggestive of a hemodynamic mechanism.32 In addition, TIAs may occur in small vessel disease; whether this represents a small thrombus in the presumed preocclusive state of a small penetrating artery is unclear.
The occurrence of a TIA is a concerning event as it may precede a stroke. In one report of over 1,700 TIAs evaluated in an emergency room, 10% went on to have a stroke within 90 days, and one-half developed within 48 hours.33 This underscores the importance of emergent evaluation and treatment of TIAs.
ACUTE ISCHEMIC STROKE: EVALUATION AND TREATMENT
It is now possible to treat selected acute strokes to reduce or abort the cerebral insult. In order to do this it is important to remember that brain tissue requires approximately 50 mL of blood flow per 100 g of tissue per minute to meet its metabolic demands. Areas with flow greater than 20 mL per 100 g/min are able to function normally, but tissue with less than 10 mL per 100 g/min promptly develops irreversible damage. Cerebral tissue with blood flow between 10 and 20 mL per 100 g/min is salvageable, if reperfused within a certain amount of time; this area is denominated the ischemic penumbra. Even without further decreases in regional blood flow, irreversible changes in the penumbra will ensue within approximately 6 hours, mainly because of excitotoxic insults arising from the core of the stroke. This damage is mediated by various neurotransmitters, mainly glutamate. This substance is normally present in the brain, but nonfunctional astrocytes are unable to recapture it, leading to high extracellular concentrations that through a number of cell-membrane interactions result in increasing intracellular calcium levels that result in cell death.34,35
The treatment of acute ischemic stroke is directed at saving the penumbra, which initially represents the major portion of the tissue at risk; over time (hours), irreversible injury develops. The three main tenets of intervention include reperfusion or opening the occluded vessel, increasing collateral flow to the ischemic penumbra, and blocking excitotoxic influnces.
REPERFUSION
Since its approval by the Food and Drug Administration (FDA), the administration of the recombinant tissue plasminogen activator (rt-PA) for intravenous (IV) thrombolytic therapy in acute ischemic stroke has become the standard of care in appropriate patients.36 The National Institute of Neurological Disorders and Stroke (NINDS) study37 showed that individuals treated with intravenous rt-PA (0.9 mg/kg of weight, not to exceed 90 mg total dose, 10% given as a bolus and the remainder over 1 hour) had one-third greater chance of being asymptomatic or with minimal sequelae than those treated with placebo. With a 6% incidence of hemorrhagic complications, it remains a potentially dangerous drug, and certain precautions should be observed to avoid a higher complication rate. Establishing the precise onset of symptoms is extremely important as r-tPA is approved for use within 3 hours of stroke onset. Administration after this period results in decreased efficacy, and possibly an elevated rate of symptomatic cerebral hemorrhage. Noncontrast brain computed tomography (CT) needs to be done urgently because the presence of cerebral hemorrhage, which cannot be reliably excluded on clinical grounds alone, precludes the use of thrombolytic medications. Patients presenting within 3 hours of onset of symptoms may have a normal brain CT, or one with early ischemic changes, such as loss of gray-white matter distinction, effacement of the insular ribbon, or loss of basal ganglia definition. A dense middle cerebral artery represents thrombus within this vessel. Further evidence of ischemia, particularly a well-established hypodensity, represents cytotoxic edema in infracted tissue, and should preclude the use of intravenous rt-PA. The brain parenchyma can also be studied anatomically by MRI. The latter is much more sensitive, particularly when diffusion sequences are obtained.38,39 Restriction of the normal diffusion of water across membranes in response to energetic failure becomes apparent within minutes and remains positive for up to 2 weeks after insult.40 Quantification of neurologic deficits is helpful in estimating the risk of administering thrombolytics and in assessing response to treatment; the National Institutes of Health Stroke Scale (NIHSS)41 is the most widely used for this purpose. An NIHSS grade greater than 24 (a higher grade indicates more deficits) is associated with more intracerebral hemorrhagic complications after therapy, but does not represent an absolute contraindication because these patients have a poor prognosis if left untreated. Conversely, minimal deficits, such as isolated sensory symptoms or an NIHSS less than 4 does not warrant the risk of thrombolytic therapy. Factors that may preclude thrombolytic use such as recent surgery, cerebral aneurysms, cranial vascular malformations or tumors, or a potential source of systemic bleeding (i.e., active gastric ulcer) should be sought. Laboratory screening for coagulopathic conditions or significant thrombocytopenia should be obtained.
Unfortunately, the 3-hour window for intravenous thrombolysis limits the number of patients treated, currently fewer than 5% of all strokes in this country. Other intravenous rt-PA studies have not shown efficacy with this agent beyond 3 hours. This has led to the exploration of intraarterial administration of thrombolytic agents, with the expectation that it may prove to be a safer and more effective way of opening occluded cerebral vessels, and may provide a longer window of opportunity for intervention than intravenous application. In PROACT II,42 prourokinase (9 mg) was administered to individuals with large strokes from a middle cerebral artery occlusion within 6 hours of onset of symptoms. There was a 15% absolute difference in improvement to complete resolution or minimal sequelae between patients treated with prourokinase (40%) versus those in the placebo group (25%). In spite of an excess in symptomatic cerebral hemorrhages in the treated group (10.2 vs. 1.8%), this did not translate into greater mortality.
It is possible that the posterior circulation has a longer window of viability than the anterior circulation.43 Small series have reported benefit from intra-arterial thrombolysis in basilar artery occlusion up to 48 hours, a condition with a mortality of up to 90% if left untreated. Brandt and colleagues43 and the AUST Study Group44 found recanalization rates of 51% to 69% with a reduction in mortality of over 50%. As the intraarterial thrombolytic process requires specialized centers and the setup of the angiographic suite is time consuming, some have attempted to initiate intravenous rt-PA early on at a reduced dose, to be followed by intraarterial thrombolysis if needed. With this approach, greater recanalization rates, compared to intravenous rt-PA, have been reported.
As diffusion-perfusion MRI and other techniques to quantify blood flow, such as perfusion CT and xenon-CT, become more widely available, clinicians will be better equipped to estimate the relative benefits and risks of thrombolysis, based more on physiologic data than rigid time constraints. Patients with large areas of penumbra tissue would benefit most, while those with large tissue volume with blood flow under 10 mL per 100 g/min would not benefit and may be at increased risk for hemorrhage. A recent report utilizing diffusion-perfusion MRI to guide thrombolysis with desmoteplase allowed the safe and apparently efficacious administration of this thrombolytic agent up to 9 hours after the onset of ischemia.45 Figure 1 exemplifies the use of diffusion-perfusion MRI in acute stroke treatment. Ideally, multimodal testing of the parenchyma to exclude hemorrhage, a viability study to determine the amount of salvageable brain, and a vascular study to assess vessel patency would provide the clinician with the information to make rational decisions regarding the need to treat. Magnetic resonance (MRI, diffusion-perfusion imaging, and magnetic resonance angiography [MRA]) and CT technology (with CT perfusion and CT angiography) have the potential to guide this multimodal evaluation.
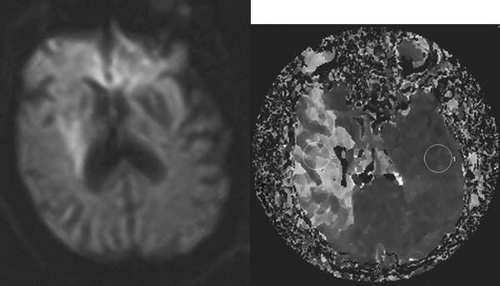 Fig. 1. An 81-year-old man with atrial fibrillation developed acute onset of left-sided weakness and presented to hospital 3 hours and 40 minutes after onset of symptoms. A: The diffusion-sequence magnetic resonance imaging (MRI), with infracted tissue in the right basal ganglia in white. B: Decreased perfusion in the right middle cerebral artery distribution (white area). Given the relatively small diffusion abnormality and the large perfusion defect, it was decided to proceed with thrombolysis despite the time elapsed. |
However, it should be noted that at the time of this writing, only intravenous rt-PA is approved by the FDA in the United States for treatment of acute ischemic stroke.
INCREASING COLLATERAL FLOW TO THE ISCHEMIC PENUMBRA
The most important single measure to reach this goal is to avoid hypotension. Because the hypertensive response after acute cerebral ischemia may be a normal compensatory response, it is recommended not to treat systolic blood pressure under 220 mm Hg or diastolic pressures below 120 mm Hg in the first 24 hours after onset of the event. If thrombolytics are used, then the systolic blood pressure should be kept under 180 mm Hg, and the diastolic BP under 110.46 In the presence of overt cardiac failure, coronary ischemia or aortic dissection, blood pressure needs to be reduced more aggressively. When blood pressure reduction is required, β-blockers such as labetalol are the preferred initial therapy. Vasodilators, particularly nifedipine, should be avoided because they can produce a precipitous drop in blood pressure resulting in reduction of cerebral blood flow to the ischemic penumbra. Patients with low blood pressures may need colloids and occasionally vasopressors to ensure adequate perfusion to the ischemic penumbra. The use of isotonic intravenous fluids is recommended unless there is a cardiovascular contraindication such as congestive heart failure.
BLOCKING EXCITOTOXIC INFLUENCES
The reduction of excitotoxic influences should preserve penumbra neurons. Multiple agents have been tried in a variety of studies with disappointing results, but the search continues.47 If a safe agent is developed, it may be administered in the prehospital setting by paramedics. Nevertheless, there are two very effective interventions with clear neuroprotective effects that should currently be used.46 The first is avoidance of hyperglycemia because it worsens ischemic injury. The second is treatment of hyperthermia: even small increases of temperature adversely affect the reversibility of ischemic penumbra damage. Fever should be treated aggressively with antipyretics and even cold blankets if needed. Hypothermia has been shown to have beneficial effects on stroke outcome in animal studies; human studies are currently underway.
ANTITHROMBOTIC THERAPY IN ACUTE ISCHEMIC STROKE
The use of antithrombotic agents is based mainly on the risk of early recurrence of ischemia, and to a lesser degree on prevention of progression of symptoms. Although earlier reports suggested a high risk of early recurrent stroke,48 most recent large studies suggest a low risk of early recurrence. In the placebo arm of early anticoagulation trials, the risk of recurrence was 1.1%49 to 4.4%50 in the first weeks after a stroke, and even in cardioembolism early recurrence is rare.51,52 In addition, full anticoagulation carries a certain risk of cerebral hemorrhage.50 As to early deterioration, there is a theoretical concern that an intravascular thrombus may extend and cause further ischemic injury. However, the causes of early progression of deficits are multiple and include cerebral edema, hemorrhagic transformation and infections, amongst others. A number of trials of very early anticoagulation with unfractionated heparin, low-molecular-weight heparins, and heparinoids have not shown any benefit of this practice in improving outcomes.46 Therefore, full anticoagulation is not indicated in the acute phase after an ischemic stroke. For most patients, antiplatelet therapy should be administered.53 However, subcutaneous anticoagulants for prevention of deep vein thrombosis are usually indicated in those with impaired mobility. After the acute period, the appropriate antithrombotic therapy is instituted based on the mechanism of ischemia.
EVALUATION OF THE CAUSES OF CEREBRAL ISCHEMIA AND PREVENTION OF RECURRENT STROKE
Once the acute stroke has been treated (or when confronted by a TIA, evidence of an old stroke, or asymptomatic arterial disease) then evaluation of the risk factors and mechanisms of ischemia is warranted in order to establish rational and effective preventive interventions. In the evaluation of stroke, it is important to evaluate the brain parenchyma, potential embolic sources (cardiac and arterial), and vascular risk factors.
The most common mechanisms of ischemia include embolism, occlusion of small cerebral arteries, hemodynamic events, and less commonly cerebral artery in situ thrombosis (caused by arteritis or a hypercoagulable condition) and cerebral venous thrombosis.
Embolism is probably the most common cause of ischemic stroke,54 and it consists of the occlusion of a cerebral vessel by particle formed upstream in the vascular tree. The common manifestations of cerebral embolism are a sudden onset of neurologic dysfunction, with deficits maximum at onset, and that commonly have associated cortical signs and symptoms. The embolic material will depend on the source of embolism; for example, thrombus formed in the heart chambers or deep veins is rich in erythrocytes and fibrin (red clot), while the material that arises from the large vessels, and that is often associated with atherosclerosis may be platelet-rich (white clot), and may also contain atherosclerotic plaque components such as cholesterol crystals, calcium and other debris. Determination of the embolic source and the probable composition of the embolic material are important to institute effective and rational therapy.
CARDIOEMBOLISM
Cardioembolism accounts for 15% to 30% of all embolic strokes.55,56 Thrombus formation is extraordinarily rare in a healthy heart, but structural changes, such as valvular diseases (particularly of the mitral valve), ventricular wall dysfunction (ventricular akinesis or aneurysm) after myocardial ischemia, and septal abnormalities such as a patent foramen ovale and atrial septal aneurysm, constitute a favorable milieu for generation of emboli.
Cardiac rhythm abnormality, particularly atrial fibrillation, is the predominant source of cardioembolism. The noncontracting and often dilated left atria and atrial appendage are nidus for thrombi, which can then be expelled into the cerebrovascular circulation. The incidence of atrial fibrillation duplicates with each decade of life, reaching a frequency of 39 per 1,000 in those aged 80 to 89.57 The risk of cerebral ischemia is much greater if there is associated valvular disease.58 When it is not, the presence of other factors such as hypertension, diabetes mellitus, heart failure, coronary artery disease, and certain echocardiographic findings such as dilated atria and “smoke” in the left atria appears to predict embolic risk.59,60,61 The risk, however, is similar for chronic and paroxysmal atrial fibrillation.62
The patent foramen ovale (PFO) is increasingly recognized as a common cause of cerebral embolism. It is formed as a channel between the septum primum and septum secundum that divide the atria. Although required during in utero life, this right-to-left atrial communication usually closes at birth with the recruitment of the pulmonary circulation. However, in up to one in four it remains patent,63 either as a constant anatomic channel or as a potential conduit that opens up with increases in right heart pressures. There are many possible mechanisms through which the PFO may contribute to cerebral embolism. The first is as a mere channel through which thrombi generated in the deep veins of the extremities or pelvis can bypass the lung filter and reach the brain. These thrombi may develop in the presence of a hypercoagulable condition such as immobility or stasis, dehydration, postoperative state, or in the presence of a congenital or acquired procoagulant factor. Although a PFO is a common finding, paradoxical embolization is relatively rare. Factors that increase the risk of stroke in the presence of a PFO include a large PFO, an associated hypercoagulable state, right atrial developmental variants such as a Chiari network and a Eustachian valve that may act as nidus for thrombi formation, and, more importantly, the presence of a floppy interatrial septum also denominated atrial septal aneurysm (ASA). The coexistence of a PFO with an ASA appears to be the most dangerous anatomic condition in predicting stroke recurrence. Mas and colleagues64 found a threefold risk of recurrence with a PFO plus an ASA compared to a PFO alone. Therefore, some have suggested that thrombosis may occur within a complex PFO itself. Finally, there appears to be some increased risk of developing atrial fibrillation in the presence of a PFO. Other conditions that may result in embolic cerebral damage and that have been associated with the PFO include the fat embolism syndrome (fat globules)65 and decompression illness (gaseous particles or bubbles).66
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


