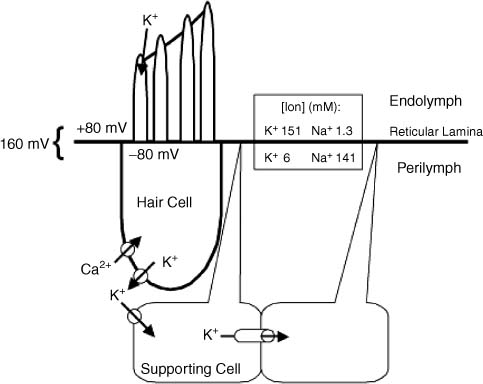
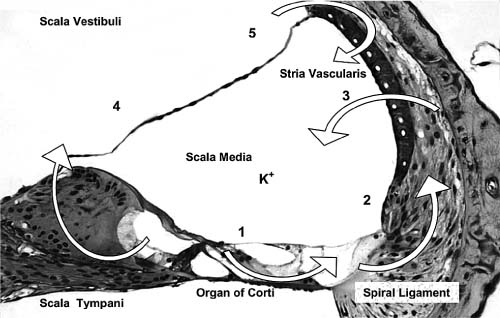
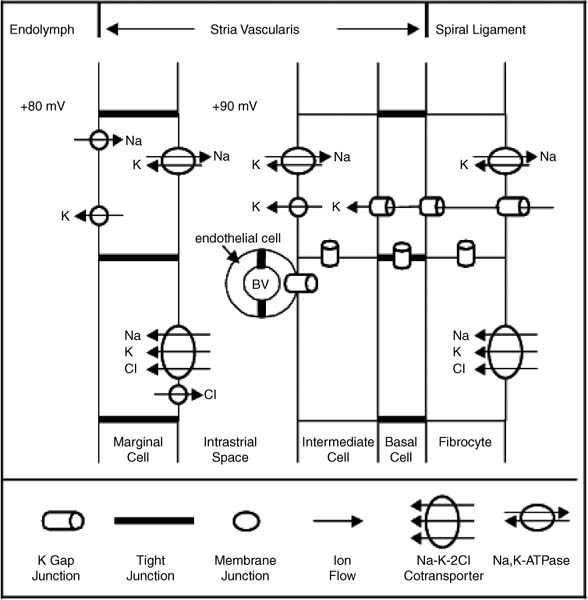
3 Ion Homeostasis and Inner Ear Disease Inner ear homeostasis refers to the processes by which the chemical equilibrium of inner ear fluids and tissues is maintained. The inner ear can function properly only if there is tight control of ion movements across its cell membranes. This includes hair cell function, regulation of extracellular endolymph and perilymph composition, and nerve impulse conductance. The major ions involved are sodium (Na+) and potassium (K+), but a significant role also is assigned to calcium (Ca2+), chloride (Cl–), and others. A thorough functional characterization of inner ear ion homeostasis is beyond the scope of this chapter and can be found in excellent reviews elsewhere.1–5 Ion homeostasis in the ear is controlled by numerous ion channels and transporters in the plasma membranes of its cells, particularly those lining the scala media. These membrane ion channels and transporters are as diverse as the ions they move, and numerous genes regulate their expression. Furthermore, inner ear ion homeostasis is dependent on the active transport of water through its own channels (aquaporins), because mere passive diffusion of water through the cell membrane is simply not sufficient to maintain the necessary osmotic pressures. The disruption of any of these channels and transporters can lead to inadequate ion transport, inappropriate fluid balances, and altered vestibular and cochlear functions. Although these inner ear channels and transporters are mainly controlled by factors in their immediate environment, many also are influenced by circulating hormones. Although this makes the ear susceptible to systemic hormone imbalances and other physiologic disorders, it also expands treatment options for dysfunctional homeostatic mechanisms. Many cochlear and vestibular disorders are the direct result of altered ion transport. As we learn more of homeostatic mechanisms in the inner ear and how they are impacted by disease, we will make more accurate diagnoses of the underlying pathology. The traditional classification of sensorineural, sudden, or rapidly progressing hearing loss is no longer adequate. These terms encompass various ear disorders ranging from permanent loss of hair cells or spiral ganglion neurons to spontaneous alterations in ion concentrations that temporarily raise hearing thresholds. These classifications do not adequately describe the diverse cochlear problems they encompass and can lead to incorrect treatment if the wrong assumption is made about the underlying pathology. Even the diagnosis of Meniere’s disease will eventually be replaced with more accurate differentiation of the relevant cochlear problem or disease process to effectively treat it. For example, it is now generally accepted that sudden hearing loss and Meniere’s disease can result from several different disease processes, many common to both. However, these disorders are treated with one or two classes of drugs (diuretics, steroids), and little further effort is made to identify the underlying homeostatic problem to determine the most effective therapy. This chapter briefly describes our current understanding of ion homeostasis mechanisms in the inner ear and how these processes are compromised by genetic and metabolic disorders to cause hearing and vestibular dysfunction. Today’s practicing otologist must have a good working knowledge of these disorders of inner ear homeostasis to differentiate the diverse causes of deafness. This will lead to more effective treatment of ear disorders with existing therapies, as well as help research otologists develop new therapeutic approaches that target the specific homeostatic problems. When the stereocilia of hair cells are deflected in the excitatory direction, a mechanoelectrical transduction current depolarizes the hair cells and initiates action potentials in the auditory or vestibular nerve. All inner ear structures and fluids are specialized for response to sound, or, in the case of the vestibular hair cells, response to acceleration or gravity. The endolymph and perilymph fluids have specific ion concentrations that must be maintained for hair cells to function with maximum sensitivity. In short, these fluids are the metabolic support system for hair cell function, similar to the way the cochlear partition traveling wave and tectorial membrane are the mechanical support system for hair cell function. Significant hearing loss occurs when either of these support systems fails. Hair cells of the inner ear operate within a unique ionic microenvironment of two distinct fluids. The hair cell apex and stereocilia are immersed in endolymph, whereas the cell body is bathed in perilymph (Fig. 3–1). The reticular lamina (apical surface of hair cells and supporting cells) separates the two fluid spaces. The endolymph (above the reticular lamina) is characterized by higher K+ and lower Na+ relative to the perilymph, thus endolymph is comparable to intracellular fluid. On the other hand, perilymph is similar to the extracellular fluid surrounding other cells throughout the body.1–5 It has been a half century since the first accurate measures were made of the ion concentrations within the endolymph and perilymph. Catherine Smith collaborated with Oliver Lowry to use his unique assay methods to measure K+, Na+, and Cl concentrations in the guinea pig ear.6 The values they obtained needed little correction by other researchers in the ensuing years with more sophisticated techniques. They also concluded that hair cells and their synapsing nerve fibers in the organ of Corti could not possibly be surrounded by endolymph, contrary to contemporary thinking. They suggested that for the nerve fibers to conduct impulses, they had to be surrounded by perilymph with its high Na+ (extracellular) concentrations. Their theory that the reticular lamina, not the basilar membrane, was the endolymph–perilymph boundary was eventually proven correct. The hair cell maintains an intracellular potential of 80 mV relative to the surrounding perilymph (extracellular) environment (Fig. 3–1). The high K+ concentration of endolymph creates an endocochlear potential of +80 mV relative to perilymph. This +80 mV endocochlear potential couples with the 80 mV hair cell intracellular potential for a differential potential of 160 mV. The vestibular system differs in that its endolymphatic potential is only +5 to +10 mV relative to perilymph, which creates a total potential difference only slightly greater than the 80 mV across the apical membrane of the vestibular hair cell. The reason for this will become clear later when the ion transport mechanisms of the cochlear and vestibular systems are compared. Figure 3–1 The ionic microenvironment of the hair cells. The hair cell has its stereocilia above the reticular lamina and soma below it. The endolymph bathing the stereocilia has a higher concentration of K+ relative to perilymph, but a lower concentration of Na+. When the stereocilia are deflected, K+ moves through stereocilia transduction channels into the hair cell to depolarize it. This opens voltage-gated K+ channels, allowing K+ to be released into the perilymph space around the base of the hair cell soma and the supporting cells. The K+ is then taken up by the supporting cells and transported toward the lateral wall and spiral ligament. The +80 mV endocochlear potential couples to the 80 mV intracellular potential of the hair cell to create the 160 mV potential gradient that drives the transduction of hair cells. (Ion values from Wangemann P, Schacht J, In: Dallos P, Popper AN, Fay RR, eds. The Cochlea. New York: Springer-Verlag; 1996:130–185.) This arrangement of endolymph and perilymph is necessary for hair cell depolarization when its stereocilia are deflected by the traveling wave. Stereocilia are laterally displaced either by shearing movement of the tectorial membrane (outer hair cells) or motion of the endolymphatic fluid (inner hair cells). Stereocilia displacement allows K+ ions to enter the hair cell through apical transduction channels, leading to its depolarization and neurotransmitter release. The flow of K+ ions is down an electrochemical gradient that brings K+ into the cell from the endolymph and eventually out the base of the cell body into the perilymph (higher K+ in hair cell than in perilymph). If the entire hair cell was surrounded by endolymph, the hair cell could not function because the K+ ion concentration inside and outside the cell would be equal. Thus, the higher K+ ion concentration around the stereocilia provides for a receptor current that does not require an energy dependent pump in the hair cell. The vestibular hair cells operate under the same endolymph–perilymph ion differential for transduction. When these two fluids are not different, transduction is compromised and hearing loss or vestibular dysfunction occurs. Such would be the case during a Meniere’s attack when rupture of the membranous labyrinth allows endolymph and perilymph to mix, essentially eliminating the endolymphatic (endocochlear) potential. The inner ear has an elaborate system by which it maintains the critical K+ and Na+ concentrations in the endolymph and perilymph.1–3,7 Following release by the hair cells, K+ is moved laterally through gap junctions between supporting cells in the organ of Corti (Fig. 3–1). When ions reach the spiral ligament, gap junctions between fibrocytes move ions up to the stria vascularis for secretion back into the endolymph (Fig. 3–2). The stria vascularis has a well-developed ion transport system of diverse channels and transporters that move K+ into the endolymph and Na+ out of the endolymph and into the perilymph. Other ions, such as Cl, H+, and Ca2+, also are actively transported into or out of the endolymph by the stria vascularis. Many of these same channels and transporters exist in hair cells and other epithelial cells around the scala media,1–3,8–10 but we will focus on the stria because it is the most complex and best characterized. Figure 3–2 Recycling of K+ in the cochlea. This mouse cochlear turn illustrates the transport of K+ ions after they are released from the hair cells by depolarization. Ions are moved through gap junctions between adjacent cells to eventually be secreted by the stria vascularis back into the endolymph. The major recycling pathway is laterally along the supporting cells (1), to the spiral ligament fibrocytes (2), where they reach the stria vascularis (3). Some K+ transport also occurs medially through the cells lining the spiral limbus (4), into the scala vestibuli (5) for transport to the stria vascularis. The major ion channels and transporters involved in stria ion movement include Na+,K+–adenosine triphosphatase (Na+,K+–ATPase),11 the Na-K-2Cl cotransporter,12,13 epithelial Na+ channel,14–16 several different K+ channels,2,17,18 Cl channel,19,20 and the K+–H+ exchanger.21 The positions of these ion channels and transporters in the stria vascularis are determined by the direction of movement of a particular ion (Fig. 3–3). Most of these ion channels and transporters also occur in secretory or absorptive tissues (salivary glands, kidney), but their relationship to the luminal or basal sides of epithelial cells varies depending on the required direction of K+ and Na+ flow. The 80 to 90 mV endocochlear potential is generated by the intermediate cells within the intrastrial space (Fig. 3–3). The marginal cells, while serving as the final transport epithelium to move K+ into the endolymph, do not appreciably alter the endocochlear potential. This minimal contribution by the marginal cells is paralleled by the minor contribution to the vestibular endolymphatic potential by the vestibular dark cells, which are the only ion transport cells operating there.22 The vestibular endolymphatic potentials are less than 10 mV because the single layer of dark cells does not have the benefit of underlying transport layers like the intermediate and basal cells of the stria vascularis. The cellular mechanism for moving K+ to the strial intermediate cells can be thought of as an epithelial complex made up of the fibrocytes in the spiral ligament, the basal and intermediate cells of the stria, and the stria capillary endothelial cells.7,18,23 All of these cells are interconnected by gap junctions, facilitating the transport of K+ between them and into the intrastrial space (Fig. 3–3). The tight junctions of the vascular endothelial cells and basal cells also contribute to the endocochlear potential by limiting intercellular leakage of ions. The exchange of ions through these cells is driven by the Na+,K+-ATPase system in the fibrocytes, intermediate cells, and marginal cells. Thus, any cochlear disorder that compromises these strial cells layers, channels, transporters, or tight junctions will reduce the endocochlear potential and cause hearing loss. Also, the ion transport components of the stria system are controlled by multiple genes, making the production of endolymph susceptible to a variety of genetic disorders. Although water easily diffuses through cell membranes, such a passive water exchange is not enough to maintain the osmotic homeostasis of the inner ear. Therefore, the ear, along with several other fluid producing organs (kidney, salivary glands), employs active water transport through aquaporin channels. To date, approximately 11 aquaporins have been identified and nearly all occur within the inner ear, particularly in areas controlling fluid homeostasis.24 Because these different aquaporins appear to subserve unique homeostatic processes, they have nonoverlapping distributions within the ear. Currently little is known about hearing loss resulting from aquaporin dysfunction, except that knockout of AQP4 leads to hearing loss.25 Viral infections are known to shut down aquaporin function in the lung to increase inflammation and edema.26 Thus, hearing loss due to viral and bacterial labyrinthitis may result from similar cochlear aquaporin dysfunction. Figure 3–3 Major ion channels and transporters in the stria vascularis. Movement of ions by the stria vascularis generates the endocochlear potential of +80 mV in the endolymph. Multiple channels are responsible for moving K+ into the endolymph and Na+ out, including the Na+,K+–adenosine triphosphatase (Na+,K+– ATPase) transport channel, the Na-K-2Cl cotransporter, and numerous membrane channels for specific ions. The fibrocytes of the lateral wall are connected by gap junctions with the basal and intermediate cells of the stria, facilitating the movement of K+ ions from the perilymph to the intrastrial compartment. Tight junctions seal adjacent marginal cells and basal cells to prevent ion leakage between cells and preserve the ionic potentials. The endothelial cells of the stria blood vessels (BV) also are sealed by tight junctions to preserve the ion potentials within the stria. These endothelial cells also are connected by gap junctions with the intermediate cell-fibrocyte complex. The +90 mV potential is initially developed within the intrastrial space, and is slightly attenuated by the marginal cells to create the +80 mV endocochlear potential (Adapted from Takeuchi S, Ando M, Kakigi A. Mechanism generating endocochlear potential: role played by intermediate cells in stria vascularis. Biophys J 2000;79:2572–2582 and Wengemann P. K+ cycling and the endocochlear potential. Hear Res 2002;165:1–9). Several ion channels and transporters within the ear are influenced by the same circulating hormones that control similar ion homeostasis mechanisms in the kidney and secretory glands. Aldosterone increases the exchange of K+ and Na+ by upregulating expression of the epithelial Na+ channel and Na+, K+–ATPase. In the kidney this leads to reabsorption of Na+ and excretion of K+. These channels in the stria move K+ into the endolymph and Na+ out of the endolymph into the perilymph (Fig. 3–3). Loss of aldosterone through adrenalectomy has been shown to reduce endolymph27 and increased aldosterone causes hydrops,28 consistent with expected K+ ion movements. Diuretics and antidiuretics have opposing effects on ion movements in the ear. Loop diuretics (ethacrynic acid, furosemide) cause stria vascularis pathology by suppressing activity of the Na-K-2Cl cotransporter and epithelial Na+ channel.29,30 Diuretics cause Na+ excretion (less absorption) in the kidney. This equates to reduced Na+ transport from endolymph to perilymph in the ear and less K+ transport into the endolymph. On the other hand, antidiuretics (vasopressin) have the opposite effect by enhancing activity of these channels to increase water and Na+ retention, working in combination with aquaporins, sodium channels, and aldosterone.31,32 In the ear, vasopressin causes hydrops,33 presumably due to increased K+ transport into the endolymph. More will be said about hormonal control of these opposing ion transport mechanisms later in the discussion of Meniere’s disease. Natriuretic peptides (atrial, C type) also are expressed in the inner ear.34,35 They are upregulated in response to increased extracellular volume, suppress vasopressin and aldosterone, and increase Na+ excretion (the same as diuretics) by decreasing Na+ and K+ exchange.36 In the ear this presumably would result in reduced strial function. It also is known that these peptides are vasoactive and influence the integrity of the blood–brain barrier, making the stria blood–labyrinth barrier a likely site of influence as well. Certainly other ion channels are at work in the inner ear and will be characterized by future research, as will the hormonal and genetic factors that control them. Nevertheless, current clinical and experimental otology research is helping to define the role of these channels and how their function is affected by various disease processes. A variety of hearing and vestibular disorders result from disruption of ion homeostasis. Gene defects in ion channels and transporters often cause permanent hearing loss, whereas many other disorders of ion transport are transient and recover, often without treatment. The disruption of stria ion transport mechanisms is the final pathway common to many of them. Cochlear ion channels and transporters can become either more active or less active, depending on the disease state or metabolic influence. As a result, hearing disorders can result from increased or decreased stria ion transport. Increased activity of the strial process can cause greater K+ transport than normal into the endolymph, leading to endolymphatic hydrops. On the other hand, dysfunction that leads to less ion transport than normal will decrease K+ transport or endolymph production, a condition of endolymphatic xerosis. It is important to understand this distinction because treatments may be different in the two conditions. Thus, the critical issue for otologists is to understand the specific ion transport dysfunction that underlies the different forms of hearing loss and determine the best therapeutic approach (if any) to restore hearing sensitivity. Identifying a patient’s condition as sensorineural hearing loss is only the beginning of the diagnostic process, not the end of it. The next discussion is a brief summary of what ion homeostatic mechanisms are likely involved in different genetic and metabolic hearing disorders. A variety of gene disorders cause permanent hearing loss due to impaired ion transport in the ear. In fact, the majority of nonsyndromic genetic hearing loss is due to altered proteins within the ion channels and transporters that prevent K+ movement from the organ of Corti, up the lateral wall, and into the stria vascularis (Fig. 3–3).37,38 An overview of the genes controlling ion transport and homeostasis shows that most regulate one particular protein that operates in a very restricted region of the ear (Fig. 3–4). For example, the genes KCNE1 and KCNQ1 each produce proteins (Isk and KvlQT1, respectively) that together make up the K+ channel on the apical membrane of the stria vascularis marginal cell. Their absence causes the lack of endolymph production and hearing loss associated with Jervell and Lange-Nielsen syndrome.2 A totally different gene is responsible for the K+ channel (KCNJ10)
Ion Homeostasis in the Ear
Na+–K+ Balances in Endolymph and Perilymph
Cochlear and Vestibular Potentials
Na+–K+ Transport Mechanisms
Aquaporins and Water Transport
Hormonal and Metabolic Control of Ear Ion Balances
Inner Ear Disorders of Ion Homeostasis
Permanent Disorders of Ion Homeostasis
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


