a codon, corresponds to a specific amino acid. Each DNA molecule is comprised of a sense or coding strand (5′ to 3′) that is paired with a complementary antisense or template strand (3′ to 5′). Segments of the DNA containing genetic information that is transcribed into corresponding RNA, proteins, and polypeptides are called genes.
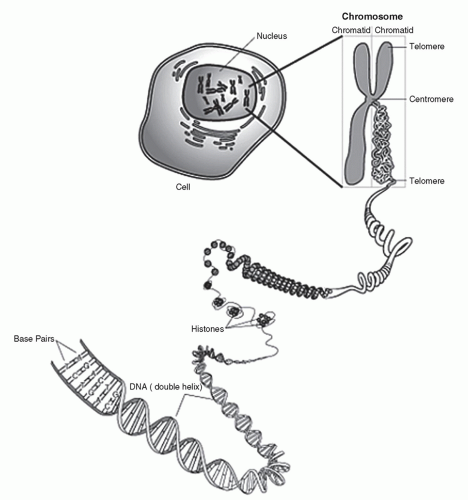 Figure 9.1 Structure and components of the chromosome. |
sperm undergo programmed destruction, leaving only the maternally mitochondria within the developing offspring (3). For this reason, mitochondrial DNA is solely of maternal origin.
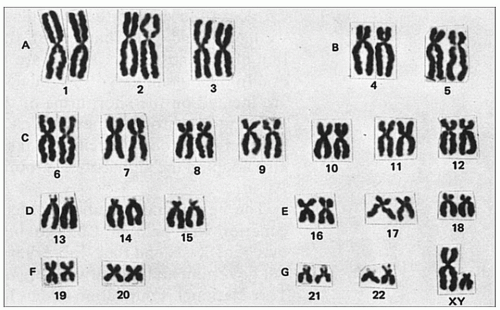 Figure 9.2 Human karyotype. |
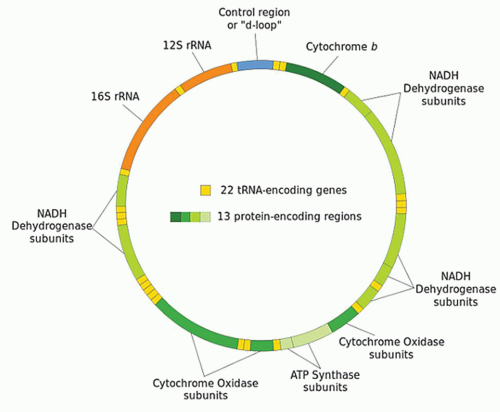 Figure 9.3 The mitochondrial genome. |
and polypeptide products (Fig. 9.4). In short, during the process of transcription, messenger RNA (mRNA) pairs with DNA, utilizing the sense strand as a template. Therefore, mRNA is configured in a 3′ to 5′ orientation and is identical to the antisense strand of DNA with the exception of the thymine to uracil substitutions. Transcription takes place completely within the cell nucleus. Once this process is complete, the mRNA exits the nucleus where, with the assistance of transfer RNA (tRNA) and ribosomal RNA (rRNA), the code is translated on a ribosome (a complex of protein and rRNA) into protein. Because the mRNA is similar to the antisense DNA strand, the nucleotide bases of the tRNA correspond directly to that of the sense DNA strand. For translation to occur, tRNAs specific for individual amino acid, with the amino acid covalently attached to it, are required. Each nucleotide triplet represents a codon, of which there are 64, corresponding to a specific amino acid or a stop signal (Fig. 9.5). The peptide produced by the tRNA ultimately forms proteins and polypeptides, which are critical to virtually every cellular function.
TABLE 9.1 TYPES OF RIBONUCLEIC ACIDS | |||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
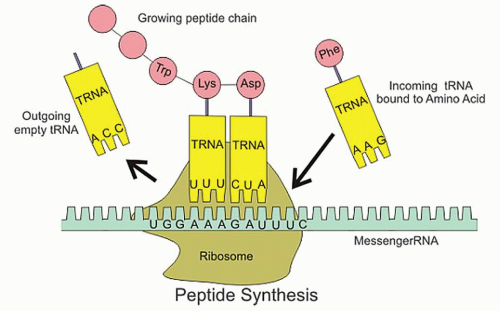 Figure 9.4 RNA translation. |
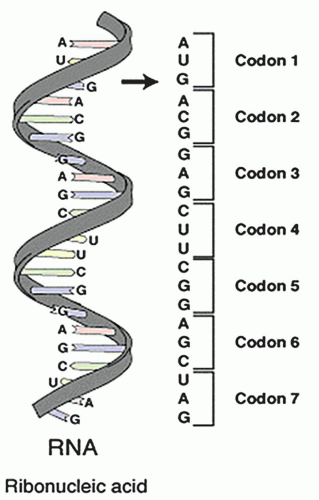 Figure 9.5 Nucleotide triplets. |
TABLE 9.2 BASIC GENETIC TERMINOLOGY | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
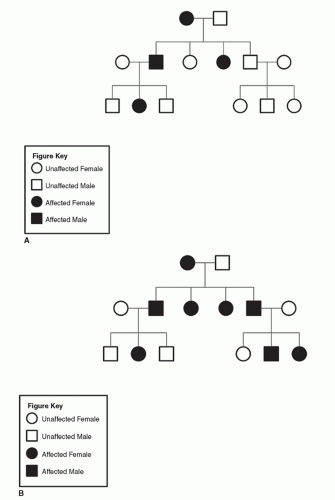 Figure 9.6 A: Pedigree of family with neurofibromatosis (heterozygous dominant parent inheritance). B: Pedigree of family with neurofibromatosis (homozygous dominant parent inheritance). |
nature owing to the normal phenotypical presentation of the carrier individuals at childbearing age. Autosomal recessive disorders with lethal phenotypes, such as Tay-Sachs disease and Bowen-Conradi syndrome, most often occur in patient with no prior family history of disease. The lethal phenotypes tend to occur more frequently among consanguineous families in which the parents are genetically related. Many autosomal recessive disorders tend to have a racial or ethnic predilection (i.e., sickle cell anemia, β-thalassemia, and Gaucher disease) believed to be a function of the founder effect. The founder effect describes the expression of a mutation initially occurring in one or few founding members of a group that has been propagated by the tendency for marriages to occur within the same group. As with autosomal dominant disorders, there is no sexual predilection associated with autosomal recessive disorders.
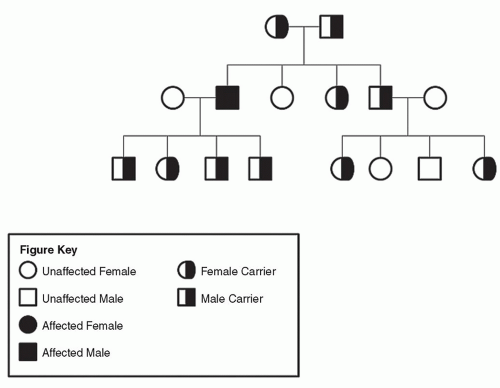 Figure 9.7 Pedigree of family with cystic fibrosis (autosomal recessive inheritance). |
inherit X-linked recessive disorders from their mother. Also, because males lack a second X chromosome, the disease allele represents the only functional X chromosome allele leading to the phenotypical expression of disease. There are a number of X-linked recessive diseases encountered in otolaryngology including X-linked stapes gusher syndrome, X-linked Alport syndrome, idiopathic hypoparathyroidism, X-linked agammaglobulinemia of Bruton, Lesch-Nyhan syndrome, severe combined immunodeficiency disease, Wiskott-Aldrich syndrome, Norrie disease, X-linked ichthyosis, Keipert syndrome (nasodigitoacoustic syndrome), and fragile X syndrome.
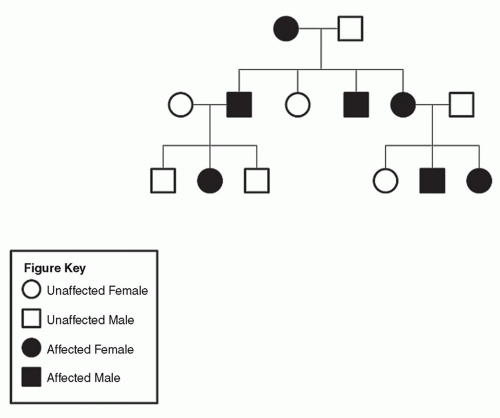 Figure 9.8 Pedigree of family with Aicardi syndrome (X-linked dominant inheritance). |
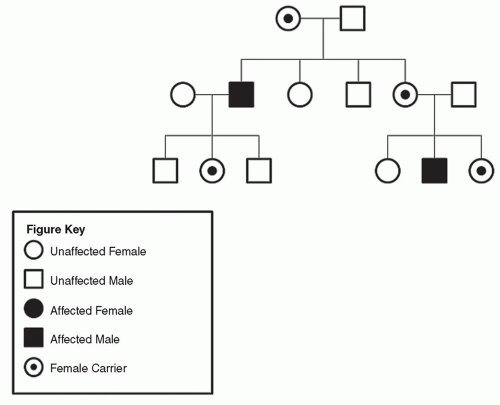 Figure 9.9 Pedigree of family with Kallmann syndrome (X-linked recessive inheritance). |
imprinting. Beckwith-Wiedemann syndrome is a disorder of genetic imprinting coincidentally described by Hans-Rudolf Wiedemann in Germany and J. Bruce Beckwith of Loma Linda University, California in the 1960s. Beckwith-Wiedemann syndrome is the most common overgrowth syndrome. It is characterized by gigantism, macroglossia, anterior abdominal wall defects (most commonly congenital exomphalos), neonatal hypoglycemia,
organomegaly, and the development of multiple tumors during childhood, most commonly Wilms tumors (16, 17, 18). H19, also known as BWS, has been found to be associated with Beckwith-Wiedemann syndrome. The paternal allele of the H19 gene is always silenced, and only the maternal allele is expressed. Interestingly, variability in H19 or BWS has been associated with tumorigenicity and variable degree of aggressive behavior of certain tumors (19, 20, 21). Similarly, the cyclin-dependent kinase inhibitor 1C gene, also known as CDKN1C or p57KIP2, has been implicated in Beckwith-Wiedemann syndrome (22). CDKN1C, like H19 undergoes the process of imprinting and only the maternal allele is expressed. CDKN1C is proposed to be a tumor suppressor gene that, when mutated, leads to the formation of various tumors (22, 23). Insulin-like growth factor 2 gene (IF2) has also been implicated in the presentation of Beckwith-Wiedemann syndrome (24). In the case of IF2, in the normal situation, only the paternal copy of the allele is expressed. In tumors derived from patients with Beckwith-Wiedemann syndrome, a lack of imprinting has been demonstrated with both alleles being expressed in association with the disease.
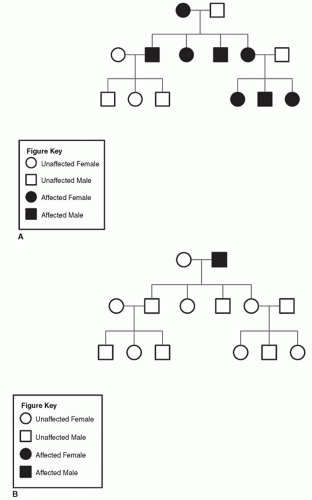 Figure 9.10 A: Pedigree of family with MINSHL (maternal inheritance). B: Pedigree of family with MINSHL (paternal inheritance). |
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


