11 Intraocular Tumors in Adults
MELANOCYTIC TUMORS OF THE UVEAL TRACT
Introduction
In adults, most intraocular tumors arise from, or involve the uveal tract, the eye’s middle coat composed of the stroma of the iris, ciliary body, and choroid. The uvea tract is highly vascular and pigmented. The pigment is contained within the cytoplasm of dendritic uveal melanocytes, which are derived embryologically from the neural crest (Fig. 11-1). A similar number of uveal melanocytes are present in lightly and heavily pigmented eyes. Increasing intensity of uveal pigmentation (and eye color) is caused by a corresponding increase in the size and number of melanin pigment granules or melanosomes in the cytoplasm of the melanocytes. The pigment may protect against the development of uveal tumors, because uveal malignant melanoma occurs most often in patients with blue eyes and is rare in heavily pigmented individuals.
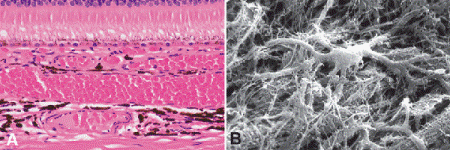
Fig. 11-1. A. Choroid. A. The choroid is the posterior part of the uveal tract. Its stroma contains dendritiform melanocytes and vessels. The latter include the choriocapillaris, which is located directly beneath Bruch membrane and the Sattler and Haller layers composed of progressively larger vessels. B. Scanning electron micrograph shows dendritic configuration of choroidal melanocyte. (A. H&E ×100, B. SEM ×1,250)
The choroidal vessels occasionally undergo hamartomatous proliferation forming hemangiomas, and the rich vascular supply of the posterior choroid explains that region’s predilection for blood-borne metastases.
NEVI
Uveal nevi are benign melanocytic neoplasms that are incapable of metastasis. Nevi, which occur in 5% of adults, are the most common intraocular tumor. Most choroidal nevi typically appear as flat or minimally elevated patches of increased choroidal pigmentation that measure 1 to 2 mm in diameter and are <2 mm in thickness (Fig. 11-2A). They may be pigmented or amelanotic, and they often have an irregular or feathery border. Drusen often develop on the surface of nevi with time. Most nevi are stationary lesions that do not change on serial observation. However, nevi occasionally do undergo malignant transformation into malignant melanoma; the rate of malignant transformation is estimated to be only 1/10,000 to 15,000 per year.
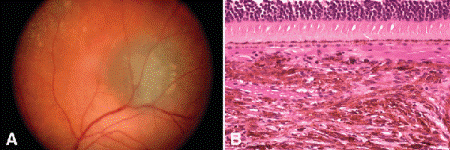
Fig. 11-2. Choroidal nevus. A. Small pigmented lesion presumed to be nevus remained stable on followup. B. Choroidal nevus. Choroid contains infiltrate of pigmented spindle cells with bland nuclei. The RPE and choriocapillaris are intact, and the overlying retina remains attached. (B. H&E ×100)
It may be difficult to differentiate between a choroidal nevus and a small malignant melanoma clinically. The observation of tumor growth may be the only clinical criterion that is helpful in this regard. Clinical factors that suggest that a pigmented lesion will grow and probably is a melanoma include the presence of symptoms, subretinal fluid and orange pigment, tumor thickness >2 mm, and contact of the posterior margin of the lesion with the optic disc. A small melanocytic lesion of the choroid with none of these factors has a 3% risk of growth into melanoma at 5 years and most likely represents a choroidal nevus. Tumors that display one factor have a 38% risk of growth, and those with two or more factors show growth in over 50% of cases. Most tumors with two or more risk factors probably represent small choroidal melanomas, and early treatment is generally indicated.
Uveal nevi are bland spindle cell tumors that comprise the benign end of the biologic spectrum of melanocytic neoplasms. Histopathologically, a compact infiltrate of slender pigmented or nonpigmented spindle cells typically replaces the choroidal stroma (Fig. 11-2B). The nevus cells have bland oval or cigar-shaped nuclei that lack nucleoli or nuclear folds and have finely dispersed chromatin. Mitotic activity is absent. In some cases, the nevus cells are plump and dendritic in shape. Intranuclear cytoplasmic inclusions are common in some cases. Foamy balloon cells that appear to be undergoing lipoidal degeneration are found in 4% of nevi. Maximally pigmented, plump, polyhedral nevus cells comprise the magnocellular variant of nevus called melanocytoma (see below).
MELANOCYTOMA (MAGNOCELLULAR NEVUS)
A melanocytoma is a characteristic type of uveal nevus composed of plump polyhedral nevus cells filled with copious quantities of maximally pigmented cytoplasm. Melanocytomas have been called magnocellular nevi. In contrast to most nevi, melanocytoma may be relatively large and may be difficult to distinguish clinically from melanoma.
Melanocytomas classically involve the optic disc but can arise from any part of the uveal tract including the iris, choroid, or ciliary body. Clinically, they are intensely pigmented and often occur in young patients. Unlike melanoma, they do not have a predilection for whites; 37% to 50% of optic disc melanocytomas have been reported in African Americans.
Melanocytomas are so intensely pigmented that they appear black on routine microscopy. The copious cytoplasmic pigmentation typically obscures the nuclei of the melanocytoma cells requiring bleached sections for proper evaluation (Figs. 11-3D and 11-4). When bleached sections are examined, the tumor cells are found to have a low nuclear/cytoplasmic ratio and bland nuclei. Nucleoli usually are inconspicuous, but there are exceptions to the rule. Melanocytomas often undergo spontaneous necrosis and typically contain pigment-laden macrophages. Extensive tumor necrosis typically is observed more often in a melanocytoma than a melanoma of comparable size, and totally necrotic melanocytomas occasionally are encountered. Melanophages released by partially necrotic iris melanocytomas can cause secondary melanocytomalytic glaucoma by physically obstructing the trabecular meshwork (Fig. 8-16). Transformation into malignant melanoma occurs rarely (1%–2%).
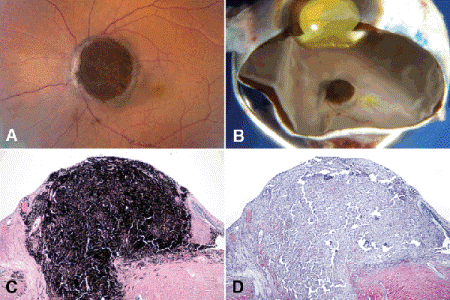
Fig. 11-3. Melanocytoma. A. Large, intensely pigmented melanocytoma of optic disc in African American woman was enucleated because patient complained of intractable pain and there was concern about malignant transformation. B. Pigmented mass protrudes from optic disc. C. Intensely pigmented tumor replaces parenchyma of optic disc. D. Bleached sections revealed no evidence of malignant transformation. (C. H&E ×25, D. Bleach ×25)
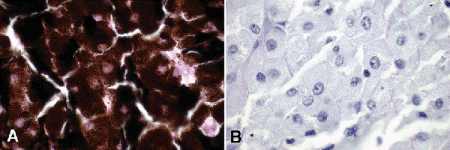
Fig. 11-4. Melanocytoma. A. Copious quantities of melanin pigment obscure nuclear details. B. Bleaching of melanin pigment discloses bland nuclei and low nuclear/cytoplasmic ratio consistent with benign magnocellular nevus. (A. H&E ×250, B. Bleach ×250)
UVEAL MALIGNANT MELANOMA
Clinical Features
Worldwide, uveal melanoma is the most common primary malignant intraocular neoplasm in adults. Uveal melanoma is the most common intraocular malignancy in the United States and Europe. Elsewhere, the pediatric retinal neoplasm retinoblastoma is more common. Uveal melanomas are relatively rare; about 1,800 tumors occur yearly in the United States. The annual age-adjusted incidence in the United States is about 6 cases per 1 million population.
Uveal melanomas arise from the dendritic melanocytes of the uvea, the middle pigmented and vascularized coat of the eye, which includes the iris, ciliary body, and the choroid (Fig. 11-1B). Choroidal melanomas are most common. Uveal melanoma affects both sexes equally. Although pediatric and even rare congenital cases have been reported, uveal melanoma generally occurs in older persons. The mean age of patients eligible for treatment in the Collaborative Ocular Melanoma Study (COMS) was 59 years. Less than 1% of cases occur in patients less than age 20 years. Older patients tend to have larger tumors and are more likely to die from their tumors after enucleation.
Race is an important predisposing factor for uveal melanoma. The tumor has a predilection for Europeans with lightly colored eyes. In the United States, the incidence of uveal melanoma in white patients is 8.5 times greater than the incidence in African Americans. The tumor is also relatively uncommon in Latin America and Asia. The incidence of uveal melanoma in the United States is 15 times greater than that in Taiwan, that is, 6 per million versus 0.39 per million.
White patients who have congenital ocular or oculodermal melanocytosis (Nevus of Ota) (Fig. 5-26) are especially at risk to develop uveal malignant melanoma. The tumor is spawned by a diffuse nevus of the uvea, which is evident clinically as hyperchromic heterochromia iridum. Affected patients often have slate-gray epibulbar pigmentation and a bluish discoloration of adnexal skin as well. It has been estimated that about one in 400 white patients with oculodermal melanocytosis will develop uveal melanoma in their lifetime. This risk is about 25 times greater than the risk in unaffected patients. Melanomas have developed in young patients with Ota nevus. Ocular and oculodermal melanocytosis are relatively common in Asians but do not appear to predispose them to melanoma.
As noted above, uveal melanomas occasionally arise from localized uveal nevi, but the estimated incidence of malignant transformation is quite low. Uveal melanoma has been reported in patients with neurofibromatosis type I and the dysplastic nevus or familial atypical mole-melanoma syndrome. Uveal melanoma can arise in patients who have a rare paraneoplastic syndrome called benign diffuse uveal melanocytic proliferation or BDUMP syndrome. The tumor’s propensity for blue-eyed individuals and the inferior exposed part of the iris suggests that exposure to ultraviolet light could be a predisposing factor.
The clinical signs and symptoms of uveal malignant melanoma depend largely on the location of the tumor and the extent of the disease when the patient initially seeks medical attention. Most posterior uveal melanomas present with painless visual loss. Visual symptoms are caused most often by serous and/or solid detachment of the retina. Other mechanisms of visual loss include physical obscuration of the fovea by overhanging tumor, cystoid macular edema, cataracts caused by expanding ciliary body tumors, and rarely vitreous hemorrhage, which usually develops when tumors erode through the retina. Melanomas occasionally are found in asymptomatic patients during routine ophthalmologic examinations. Iris melanomas may present as an enlarging pigmented blemish or a change in eye color (heterochromia iridum) (Fig. 8-20). Other tumors present with unilateral glaucoma. Anterior segment melanomas cause secondary glaucoma by directly seeding or infiltrating the aqueous outflow pathways. Posterior segment tumors usually cause secondary closed-angle glaucoma through a pupillary block mechanism or by stimulating iris neovascularization. Infarcted or extensively necrotic tumors can cause prominent inflammatory signs that can mimic orbital cellulitis. Advanced cases with extrascleral tumor extension into the orbit may present with ocular proptosis. Unsuspected melanomas occasionally are found when blind, painful, glaucomatous eyes with opaque media are examined pathologically. Before the advent of ultrasonography, as many as 10% of blind, painful eyes were said to harbor previously undiagnosed tumors. Care should be taken to exclude the presence of an occult tumor preoperatively if ocular evisceration is planned. Distant metastases usually are not evident when the tumor is first detected and treated.
Diagnosis
Many posterior uveal melanomas are diagnosed by direct ophthalmoscopic visualization of the tumor by experienced clinicians (Fig. 11-5). Adjunctive studies including A and B scan ultrasonography, intravenous fluorescein angiography (IVFA), and computed tomography or magnetic resonance imaging frequently are used to confirm the clinical impression and may be particularly important if the ocular media are opacified. Transvitreal fine needle aspiration biopsy (FNAB) performed under direct ophthalmoscopic visualization occasionally is performed if the diagnosis remains uncertain after routine tests, and choice of therapy requires an accurate diagnosis. Examples where FNAB might be used include the patient who has a history of breast cancer who presents with a solitary amelanotic choroidal tumor that could be a second primary amelanotic melanoma or the patient who is thought to have a choroidal metastasis but has no history of cancer.
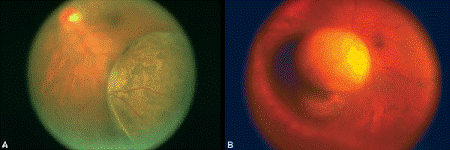
Fig. 11-5. Choroidal melanoma. A. Fundus photograph shows mushrooming head of pigmented choroidal tumor elevating inferotemporal retina. B. Tumor seen in wide angle photograph has broken through Bruch membrane and assumed characteristic mushroom or collar button configuration.
Gross Pathology
Melanomas initially arise in the uveal stroma. In early cases of choroidal melanoma, the profile of the sectioned tumor is oval or almond-shaped, and its tissue usually appears relatively cohesive after fixation (Fig. 11-6A). Although some melanomas diffusely infiltrate the uvea, most uveal melanomas are relatively well-circumscribed tumors with distinct margins. In many cases, the growing melanoma perforates Bruch membrane and enters the subretinal space where its apex typically assumes a spherical shape that often is likened to a mushroom or collar button (Figs. 11-5, 11-6B,D, and 11-7). If a choroidal tumor has a mushroom configuration, one can be reasonably certain that it is a uveal melanoma. There are exceptions to this rule, but they are exceedingly rare. Dilated blood vessels typically are found in the mushrooming head of the tumor (Fig. 11-7). The ruptured ends of Bruch membrane exert a compressive cinchlike effect on the waist of the tumor causing vascular congestion in its apex. Rupture of Bruch membrane was present in 87.7% of 1,527 large or medium sized melanomas examined in the COMS study. Retinal invasion was present in nearly half (49.1%) and tumor cells were found in the vitreous body in one quarter. About 3% of melanomas diffusely thicken the choroid without forming an elevated mass. These diffuse melanomas usually are of mixed cell type, are more likely to infiltrate the sclera, and invade the optic nerve or orbit (Fig. 11-8D). Delayed diagnosis or misdiagnosis is common.
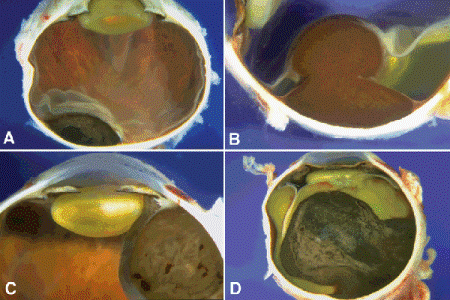
Fig. 11-6. Gross pathology, uveal melanoma. A. Small choroidal tumor has oval or almond configuration. Bruch membrane is intact. B. Heavily pigmented melanoma has arisen from the equatorial choroid and ruptured through Bruch membrane. The tumor has a characteristic mushroom or collar button configuration. Infiltration of the retina is seen at the tumor’s apex. Posterior to the tumor, the retina is detached by serous fluid. C. Ciliary body melanoma. Heavily pigmented ciliary body tumor deforms and displaces lens. D. Large choroidal melanoma. Large, heavily pigmented mushroom-shaped melanoma has produced a total retinal detachment and secondary glaucoma.
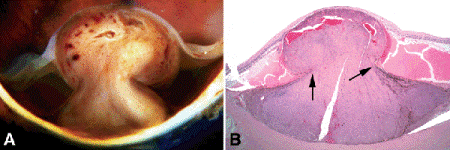
Fig. 11-7. A. Dilated vessels are present in mushrooming head of choroidal melanoma, which has ruptured through Bruch membrane. Scleral infiltration is present at the base of the tumor. The adjacent retina is detached, and the retina on the tumor’s apex is severely atrophic. B. Arrows point to edge of rupture in Bruch membrane. Dilated vessels in mushrooming head of tumor are caused by the compressive cinchlike effect of the ends of Bruch membrane on the waist of the tumor. The retina is detached by serous fluid. (B. H&E ×5)
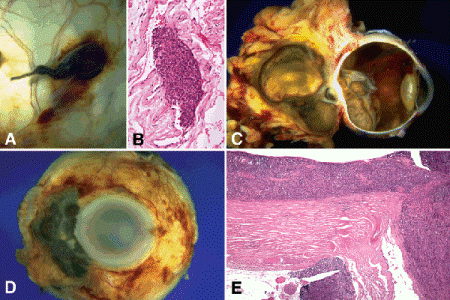
Fig. 11-8. Extraocular extension, uveal melanoma. A. Vortex vein invasion by uveal melanoma. The vortex vein in the macrophoto is massively distended by heavily pigmented tumor. B. Photomicrograph shows melanoma in the lumen of the vessel. C. Massive posterior extrascleral extension. Posterior choroidal melanoma has extended extrasclerally forming large pigmented orbital mass that dwarfs the intraocular tumor. Optic nerve invasion is present. The patient presented with ocular proptosis. D. Anterior extraocular extension. Ciliary body melanoma exited globe via anterior emissarial canals. E. Diffuse choroidal melanoma. Melanoma diffusely thickens choroid. Highly aggressive tumor has invaded optic nerve and grown through posterior emissarial canal forming juxtapapillary epibulbar mass. (B. H&E ×100, E. H&E ×25)
Choroidal melanomas are classified as small, medium, and large based on the tumor’s largest basal diameter (LTD). Small choroidal melanomas measure ≤10 mm in LTD and appear as a focal discoid or oval area of choroidal thickening. Medium-sized melanomas measure 11 to 15 mm, and large tumors are more than 15 mm in largest basal tumor diameter.
Choroidal melanomas typically cause an exudative serous detachment of the overlying and adjacent retina. Large tumors may cause total retinal detachment (Figs. 11-6B,D and 11-7). The detached retina typically shows photoreceptor atrophy and microcystoid retinal degeneration. The retinal pigment epithelium (RPE) on the surface of the tumor undergoes atrophy and proliferation forming drusenoid material and occasionally a plaque of metaplastic fibrous tissue. Many tumors infiltrate the overlying retina. The melanoma may perforate the retina in exceptional cases, causing vitreous hemorrhage and tumor seeding of the vitreous and the inner retinal surface. Large tumors may totally fill the globe. Eventually, some melanomas extend extraocularly through the sclera and invade the orbit (Fig. 11-8C,E). Secondary glaucoma is often present in eyes with advanced or neglected tumors. Uveal melanomas vary markedly in their pigment content. Some tumors are totally amelanotic. Other maximally pigmented tumors appear jet-black grossly and must be bleached before they can be interpreted histopathologically. Varying degrees of pigmentation are typically found within a single tumor. The cut surface of some tumors has a marbleized appearance. Clumps of orange pigment are found on the surface of many melanomas (Fig. 11-9). The orange pigment comprises aggregates of macrophages that have ingested lipofuscin pigment and melanin from the damaged RPE (Fig. 11-9C,D). The pigment generally is thought to be a clinical marker for an actively growing tumor and can be highlighted with fundus autofluorescence.
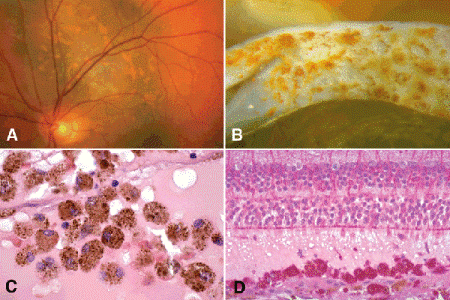
Fig. 11-9. Orange pigment. A. The presence of orange pigment is one of the factors that predicts that a pigmented choroidal lesion will grow and probably is a melanoma. B. Clumps of orange pigment adhere to posterior surface of detached retina overlying actively growing melanoma. C. Orange pigment is composed of aggregates of macrophages that have phagocytized lipofuscin and melanin pigment released by RPE cells that have been disrupted by an actively growing tumor. D. Macrophages that have ingested lipofuscin pigment are PAS-positive. (C. H&E ×250, D. PAS ×100)
Ciliary body melanomas are less common than choroidal tumors and tend to have a more spherical shape (Fig. 11-6C). Ciliary body tumors may be larger when they are first detected because they remain hidden behind the iris and often remain asymptomatic because they cause late retinal detachment. Ciliary body melanomas can deform the crystalline lens and cause unilateral cataract. Occasionally, they can invade the anterior chamber and present with iris heterochromia and secondary glaucoma. The glaucoma is caused by seeding of the trabecular meshwork by tumor cells, or by circumferential tumor growth around the angle (ring melanoma).
Histopathology
The biologic spectrum of uveal melanoma cells comprises bland spindle A melanoma cells at one end and wildly anaplastic epithelioid cells at the other (Figs. 11-10 and 11-11). The term spindle cell is derived from the fusiform or spindled configuration of the cells’ cytoplasmic outline. Spindle cells are bipolar in shape and many have long tapering processes that occasionally are visible when individual pigmented cells are seen in a largely amelanotic tumor. Spindle cells grow in a syncytial fashion and form interweaving fascicles of parallel oriented cells (Fig. 11-10A,B). The cells can be pigmented or nonpigmented.
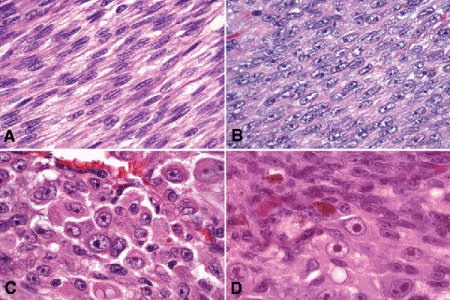
Fig. 11-10. Melanoma cells. A. Spindle A melanoma cells. Many of the cells in the photo have spindle A nuclear characteristics. The nuclei are bland slender and cigar-shaped and have finely dispersed chromatin and indistinct nucleoli. Longitudinal folds in the nuclear membrane are apparent microscopically as a chromatin stripe or line. The spindle cells form a syncytium with indistinct cytoplasmic borders. B. Spindle B melanoma cells. Most of the cells in this field are spindle B melanoma cells. They have oval nuclei and an obvious nucleolus. Compared to spindle A cells, their chromatin is more coarsely clumped. The spindle cells form a syncytium. C. Epithelioid melanoma cells. The cytoplasmic margins of these large, poorly cohesive epithelioid melanoma cells are easily discernible. Epithelioid cell nuclei are typically round and have peripheral margination of coarsely clumped chromatin. Epithelioid cells usually have prominent reddish purple nucleoli. They typically are polyhedral in shape and have copious amounts of cytoplasm. D. Uveal melanoma, mixed cell type. Mixed cell melanomas are composed of a mixture of spindle (above) and epithelioid cells (below). (All figures H&E ×250)
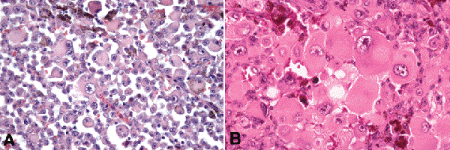
Fig. 11-11. A, B. Tumor giant cells, uveal melanoma. Tumor giant cells are highly anaplastic epithelioid cells. They are relatively rare, and their prognostic significance is uncertain. (A. H&E ×50, B. H&E ×100)
There are two types of spindle cells, spindle A and spindle B, which are distinguished by their nuclear characteristics. Spindle A nuclei are cigar-shaped and have finely dispersed chromatin (Fig. 11-10A). Many spindle A cells have a longitudinally-oriented chromatin stripe caused by a fold in the nuclear membrane. If a nucleolus is present, it usually is inconspicuous. The nuclei of spindle B cells tend to be plumper and more oval in shape and have coarser chromatin and distinct nucleoli (Fig. 11-10B).
Epithelioid melanoma cells comprise the poorly differentiated end of the cytologic spectrum. Uveal melanomas that contain epithelioid cells have a poorer prognosis. The term epithelioid, which means “epithelial-like,” reflects the superficial resemblance of the tumor cells to simple epithelial cells. Epithelioid cells have abundant cytoplasm and are often polygonal in shape (Fig. 11-10C). They have distinct cytoplasmic margins, are poorly cohesive, and do not grow as a syncytium. The nuclei of epithelioid cells typically are round or oval in shape, and they often appear vesicular due to margination or clumping of chromatin along the inner side of the nuclear membrane. Epithelioid melanoma cells also have prominent nucleoli that are often large and reddish-purple in color. The large nucleoli are often visible at lower magnification. Variants of epithelioid cells include wildly anaplastic tumor giant cells (Fig. 11-11) and relatively uniform small epithelioid cells (Fig. 11-12A).
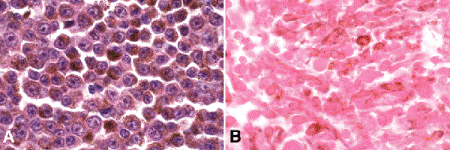
Fig. 11-12. A. Small epithelioid cells. Although these cells are relatively small, they are definitely epithelioid in character. They are polyhedral in shape and have distinct cytoplasmic outlines. The round or oval nuclei have prominent nucleoli. Clones of small epithelioid cells are encountered occasionally in uveal melanomas. B. Necrotic uveal melanoma. The cells of this necrotic melanoma are eosinophilic because they have lost their basophilic nuclear DNA. The cell type of a necrotic melanoma often can be ascertained if careful microscopy with an oil-immersion lens is performed. Necrotic uveal melanomas tend to behave clinically like mixed-cell type melanomas. (A. H&E ×250, B. H&E ×250)
During histopathologic assessment, melanoma cells are classified by their nuclear characteristics. Spindle-shaped cells that have epithelioid nuclei occasionally are encountered; such cells are classified as epithelioid. In recent years, the term intermediate cell has been used more and more. Intermediate cells are cells that have nuclear characteristics that are intermediate between spindle B and epithelioid. For example, one might apply the term intermediate cell to a spindle B cell that has a nucleus that is somewhat large and has a fairly prominent nucleolus.
Occasionally, spindle cells in a uveal melanoma are arranged radially around vessels or perpendicular to fibrovascular septa (vasocentric pattern), or their nuclei form rows that resemble the Verocay bodies or the Antoni A pattern seen in Schwannoma (Verocay pattern). Melanomas are called fascicular if these patterns dominate (Fig. 11-13A). Fascicular melanoma was a separate category in Callender’s initial classification that was dropped from McLean’s 1983 modification.
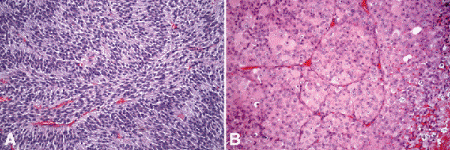
Fig. 11-13. A. Fascicular melanoma. The fascicular category of uveal melanoma has been removed from the modern revision of the Callender classification because cellular arrangement does not appear to affect prognosis. This amelanotic melanoma has a striking fascicular appearance. The nuclei of its constituent spindle cells form rows that resemble the Antoni A pattern seen in schwannoma. B. Vascular mimicry patterns, uveal melanoma. Fibrovascular septa divide parts of this predominantly epithelioid melanoma into roughly circular zones initially called vascular loops. Vascular networks are composed of adjacent vascular loops. Uveal melanomas that contain vascular mimicry patterns called loops and networks have a poorer prognosis. (A. H&E ×50, B. H&E ×50)
Varying degrees of necrosis may be are found (Fig. 11-12B). Necrosis tends to be more prominent in rapidly growing high-grade tumors, or tumors that have had prior brachytherapy. The necrosis may be patchy and focal or may involve extensive parts, or even all of the tumor. Aggregates of melanophages typically are found in the necrotic areas. Total infarction of the tumor (and other intraocular structures) may occur in eyes with severe secondary closed-angle glaucoma. As mentioned above, melanocytoma is especially prone to spontaneous necrosis. The latter diagnosis should always be considered when a totally necrotic, heavily pigmented tumor is found.
Choroidal melanomas produce abnormalities in the overlying retinal pigment epithelial including atrophy, hyperplasia, and the formation of drusen and drusenoid material. The overlying retina often shows photoreceptor loss and may develop cystoid edema. The latter tends to be more common over slower growing lesions, especially choroidal hemangiomas. After Bruch membrane has ruptured, the vessels located in the mushrooming head of the tumor are often quite prominent, reflecting vascular stagnation caused by the compression at the waist of the tumor (Fig. 11-7). Aggregates of macrophages that have ingested periodic acid-Schiff (PAS)-positive lipofuscin pigment, and melanin from the damaged RPE can be found in the subretinal fluid (Fig. 11-9). These are evident ophthalmoscopically as clumps of orange pigment that serve as a clinical marker for an actively growing neoplasm.
Uveal melanomas are placed into four categories based on their cytology. Tumors composed entirely of spindle A cells or even blander nevus cells are classified as spindle cell nevi. Tumors composed of a mixture of malignant spindle A and spindle B cells are called spindle melanomas. Melanomas of mixed cell type contain a mixture of spindle and epithelioid melanoma cells (Fig. 11-10D). Some laboratories specify the predominant cell type found in a mixed cell melanoma, for example, reporting mixed cell, predominantly spindle if only a few epithelioid cells are present. Epithelioid melanomas are composed predominantly of epithelioid cells. They are relatively rare and have the poorest prognosis. Most medium and large-sized melanomas contain a mixture of spindle and epithelioid cells. About 86% of the posterior melanomas in the COMS histopathology study were classified as mixed cell type; 8% were of spindle cell type, and 5% were epithelioid. The association between cytology and mortality is known as the Callender classification (see section on prognostic factors below).
Prognostic Factors
About one half of patients with choroidal and ciliochoroidal malignant melanomas eventually die from their tumors. Because the eye and orbit lack lymphatics, uveal melanoma spreads via the blood stream. Hematogenous metastasis to the liver occurs most often; more than 90% of cases with metastatic melanoma have liver metastases, and they are the first metastases detected in 80% (Fig. 11-14). For this reason, liver enzymes and hepatic imaging are used clinically to monitor patients for recurrence. Other common sites of metastatic uveal melanoma include the lung (24%) and bone (16%). Multiple sites are found in 87%. Unfortunately, once distant metastases are manifest clinically, therapy generally is ineffective. More than 50% of patients who have metastatic uveal melanoma die within 1 year. In recent years, there has been an effort to identify prognostic factors that could identify patients at high risk for metastatic disease who hopefully might benefit from prophylactic chemo- or immunotherapy.
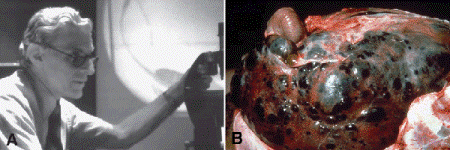
Fig. 11-14. A. Dr. Lorenz E. Zimmerman conducting ophthalmic pathology conference at the Armed Forces Institute of Pathology in Washington, DC circa 1978. A short time later “Zimm” formulated his hypothesis concerning the effect of enucleation on the dissemination of uveal melanoma. B. Liver metastases, uveal melanoma. Postmortem examination of patient who died from metastatic uveal melanoma shows massive replacement of liver by tumor. (Slide courtesy of Dr. Daniel M. Albert.)
The association between the cytologic characteristics of uveal melanoma (“cell type”) and mortality was initially reported in 1931 by Major George Russel Callender who examined a series of cases on file in the Registry of Ophthalmic Pathology at the Army Medical Museum in Washington, DC. Callender observed that melanomas were composed of two types of spindle cells that he designated spindle A and B and less differentiated epithelioid cells. He found that tumors that contained epithelioid cells had a poorer prognosis. Ian McLean et al. at the Armed Forces Institute of Pathology modified Callender’s original classification in 1978.
The presence or absence of epithelioid cells is an extremely important prognostic factor in uveal melanoma. McLean reviewed a series of 3,432 cases of malignant melanoma of the choroid and ciliary body on file in the AFIP’s Registry of Ophthalmic Pathology and found that 56% were mixed cell tumors composed of a mixture of spindle and epithelioid cells. The 15-year mortality of patients with melanomas of mixed cell type was three times that of patients whose tumors were composed solely of spindle cells. Tumor size, measured as the largest tumor diameter (LTD), was also highly correlated with mortality.
Callender cell type as modified at the Armed Forces Institute of Pathology remains one of the most reliable prognosticators of mortality from uveal melanoma. Tumor mortality is greater if uveal melanomas contain epithelioid cells (epithelioid melanomas or mixed epithelioid/spindle cell type). The 5-year mortality of uveal melanomas that contain epithelioid cells is 42%. At 15 years, death from metastatic melanoma increases to 63%. The prognosis of spindle cell tumors is much better; 90% survive 5 years and 72% survive 15 years. Although rare fatal spindle A melanomas have been reported, most tumors composed entirely of spindle A cells are thought to be benign spindle cell nevi.
Cell type remains one of prognostic mainstays of surgical pathologists because assessment is relatively rapid and requires no special stains or equipment. However, determination of cell type is highly subjective and diagnostic accuracy of can vary with the expertise and experience of the pathologist. A masked study showed that even experienced ophthalmic pathologists disagree about their classification of individual tumor cells. Secondly, melanoma cells constitute a continuous biologic spectrum that includes extremely bland spindle A melanoma cells at one end and highly anaplastic epithelioid cells at the other. Despite this, only three categories—spindle, mixed or epithelioid—are available for the classification of a given tumor, and tumors in a single category, for example, mixed cell type, can vary significantly in their apparent degree of differentiation.
The limitations of the Callender classification prompted a search for more objective and reliable criteria for the histopathologic assessment of the malignant potential of uveal melanomas. Gamel et al. showed that certain nucleolar parameters, most notably the inverse of the standard deviation of the area of the nucleolus, were useful predictors of death from metastatic melanoma. Gamel’s laboratory subsequently developed another simpler objective method of nucleolar assessment based on the measurement of the ten largest nucleoli. Although these techniques more accurately predicted survival after enucleation using morphologic data contained within routine histologic slides, they were not widely adopted because they were labor-intensive and relatively time-consuming and required special expertise and equipment.
Other attempts to make the assessment of cell type more objective and quantitative include counting the number of epithelioid cells and intermediate cells in 40 high power fields. The mitotic activity of uveal melanoma is routinely assessed by counting the number of mitotic figures in 40 high power fields.
Tumor size is another important prognostic factor. Large melanomas have a poorer prognosis than medium and small-sized melanomas. Tumor size generally is recorded as the largest tumor diameter or LTD measured at the base. The 5-year survival of small (<10 mm), medium (10–15 mm), and large (>15 mm) melanomas are 86%, 66%, and 56%, respectively. These survival rates drop to 76%, 51%, and 41% at 10 years and 70%, 43%, and 35% at 15 years. Smaller melanomas are more likely to be spindle cell tumors.
Certain extracellular matrix patterns within uveal melanomas that initially were termed microvascular patterns have been shown to be prognostic indicators for death from metastatic melanoma. The so-called vascular loops and networks composed of back-to-back loops encircling microdomains of tumor are the vascular mimicry patterns that are strongly associated with death from metastatic melanoma (Fig. 11-13B). The term vasculogenic mimicry looping matrix patterns has been applied to these patterns in recent publications.
Other prognostic factors, which have been shown by multivariant statistical analysis to be less important, include mitotic activity, extraocular tumor extension (Fig. 11-8), necrosis, pigmentation, anterior location, and lymphocytic infiltration. Paradoxically, the prognosis of heavily pigmented tumors may be slightly poorer.
Ocular pathologists routinely assess the mitotic activity of uveal melanoma by counting the number of mitotic figures in 40 high power (“high dry”) microscopic fields. Forty fields are counted because most uveal melanomas contain relatively few mitoses, that is, only five or ten per 40 HPF. Not unexpectedly, patients whose tumors have more mitoses have a poorer prognosis.
About 8% of 1,527 enucleated globes with uveal melanoma evaluated in the COMS had some degree of extrascleral extension on histopathologic examination. Although direct scleral infiltration occurs in some cases, melanomas typically extend out of the eye through the emissarial canals of vessels and nerves in the sclera, or via the lumina of the vortex veins (Fig. 11-8). Unlike retinoblastoma, uveal melanoma rarely invades the optic nerve. Coupland and Damato found that extraocular spread correlates with increased mortality because it is associated with increased tumor malignancy and, in the case of posterior tumors, more advanced disease.
The presence of tumor infiltrating lymphocytes is associated with decreased survival. De la Cruz et al examined 1,078 cases of uveal melanoma with known survival and found that 12.4% harbored 100 or more lymphocytes per 20 high-power (×400) microscopic fields. The survival rate at 15 years was 36.7% for patients in the high lymphocytic group and 69.6% for patients in the low lymphocytic group. This seemingly counterintuitive observation is explained by the fact that extraocular dissemination of tumor cells is a requisite for stimulation of a T lymphocyte-mediated immune response.
Uveal melanomas harbor recurrent nonrandom chromosomal abnormalities that include monosomy 3, trisomy 8, and structural or numerical abnormalities of chromosome 6. Loss of chromosome 3 and gains in chromosome 8 are associated with metastatic death. Monosomy 3 has been shown to be a significant predictor of poor prognosis in uveal melanoma. In one study, 57% of patients with monosomy 3 had developed metastases at 3 years, compared to none of the patients wth disomy 3. Chromosomal 3 abnormalities have been identified using a variety of techniques including fluorescence in situ hybridization and DNA amplification and microsatellite assay. Increasingly used in recent years, chromosomal analysis enables clinicians to reassure patients with a good prognosis and identifies high-risk patients who need intensive systemic screening.
Microarray analysis has also been used to assess gene expression profiling in primary uveal melanoma. Harbour et al. showed that uveal melanomas naturally cluster into two groups based on their gene expression signature. This gene expression–based classification appears to accurately predict metastatic death. Class I melanomas are low-grade and do not metastasize. In contrast, patients who have class 2 tumors are at high risk for metastases. Class II tumors have a primitive neural/ectodermal stem cell–like phenotype and are more likely to contain epithelioid cells, looping extracellular matrix patterns and monosomy 3. They are characterized by down-regulation of neural crest and melanocyte-specific genes and up-regulation of epithelial genes. Molecular classification based on gene expression profiling of the primary tumor has been reported to be superior to monosomy 3 and clinicopathologic prognostic factors for predicting metastasis in uveal melanoma. The technique is commercially available.
IRIS NEVUS AND MELANOMA
Most melanocytic lesions of the iris are benign nevi or low-grade spindle cell tumors (Figs. 11-15 and 11-16). About half of the adult population has small nonprogressive iris nevi called freckles (Fig. 11-15). Larger pigmented iris lesions initially should be observed for growth. Only 6.5% will enlarge during a 5-year observation period. Clinical features that suggest that a pigmented iris tumor is a melanoma include large size, documented growth, elevated intraocular pressure, hyphema, and tumor vascularity. The mean age of patients with iris melanomas is about 10 years younger (age 43 years) than the age of patients with posterior segment melanomas. The prognosis of iris melanoma is also relatively favorable compared to tumors of the posterior segment. Shields studied 169 patients with histologically confirmed iris melanoma and found that distant metastases developed in 5% at 10 years follow-up. The relatively small size of most iris melanomas probably is a major factor in their good prognosis.
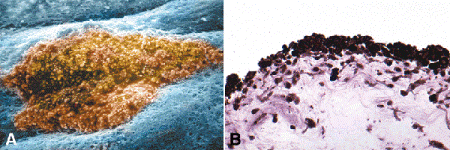
Fig. 11-15. Iris freckle. A. Sharply delimited colony of nevus cells in scanning electron micrograph is located anterior to the plane of the surrounding iris stroma. Round cellular processes decorate surface of nevus. B. Iris freckles are small nonprogressive nevi that occur in nearly half the adult population. Their constituent cells have rounded cellular processes that are densely packed with large melanosomes. (A. False-colorized scanning electron micrograph ×160 [Modified from Eagle RC Jr. Congenital, developmental and degenerative disorders of the iris and ciliary body. In Albert DM, Jakobiec FA, eds. Principles and Practice of Ophthalmology. Clinical Practice, vol. 1. Philadelphia, PA: Saunders, 1993:367–389], B. Epon section, PD ×100)
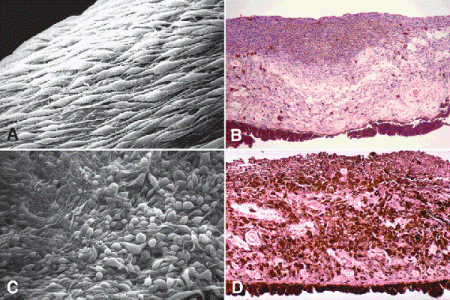
Fig. 11-16. Iris melanoma. A. Spindle cells with long tapering processes cover surface of iris melanoma. B. Low-grade spindle cell melanoma of the iris. Moderately pigmented spindle cells infiltrate and thicken stroma and form plaque on anterior iridic surface. The nuclei of the tumor cells are quite bland. C. Mixed cell melanoma of iris. Scanning electron microscopy discloses bizarrely pleomorphic cells presumed to be epithelioid cells on surface of tumor. D. Mixed cell melanoma of iris. The heavily pigmented tumor is composed of a mixture of spindle and epithelioid melanoma cells. The epithelioid cells in the stroma are distinguished by their round cytoplasmic profiles and round nuclei with prominent nucleoli. The tumor cells form a plaque on the anterior surface of the iris. (A. SEM ×640, B. H&E ×50, C. SEM ×320 [Modified from Eagle RC Jr. Iris pigmentation and pigmented lesion: an ultrastructural study. Trans Am Ophthalmol Soc 1989;87:581–687], D. H&E ×50)
Diffuse iris melanomas that cause hyperchromic heterochromia iridis and secondary glaucoma are a rare but clinically important group of iris tumors (Fig. 8-19). Many diffuse iris melanomas are higher grade tumors that contain epithelioid cells, which are poorly cohesive and prone to aqueous dispersal. Patients are often misdiagnosed clinically and undergo filtering surgery for glaucoma. The latter invariably fails and puts patients at greater risk for extraocular extension and metastasis. Pigmented tumors of the iris pigment epithelium are exceedingly rare (Fig. 11-17).
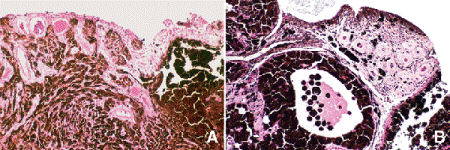
Fig. 11-17. Adenoma of iris pigment epithelium. A. Cords and sheets of intensely pigmented epithelial cells comprise rare iris tumor. B. Islands of heavily pigmented iris pigment epithelial cells erode through iris and invade anterior chamber. (A. H&E ×50, B. H&E ×100)
Treatment
Current therapy for uveal melanoma is unsatisfactory in many cases. Although metastatic disease typically is not evident clinically when the patient presents to the ophthalmologist, it currently is believed that many uveal melanomas continuously shed tumor cells into the circulation and that systemic micrometastases are present several years before the tumor is treated. Therefore, in many instances, the ophthalmologist merely achieves local control. What he or she does ultimately has little effect on systemic spread or surivival. To make matters worse, current chemotherapeutic regimens have little effect on metastatic uveal melanoma; more than 50% of patients die within 1 year.
Local tumor control is achieved in the majority of cases by enucleation or radiotherapy, typically plaque brachytherapy. The latter treatment employs radioactive plaques composed of radioisotopes such as iodine-125. The plaques are surgically affixed to the sclera external to uveal tumors and left in place for period of time calculated to deliver a lethal dose of radiation to the tumor. Radiation therapy with beams of protons or helium ions is performed at a few centers. Larger tumors or tumors that have caused secondary glaucoma generally are enucleated. Medium sized tumors can be treated with brachytherapy. The COMS showed that the mortality rates after I-125 plaque therapy and enucleation are similar. Another arm of the COMS study showed that pre-enucleation external beam radiotherapy of large choroidal melanomas does not improve survival. The COMS study also shed doubt on Zimmerman’s hypothesis that enucleation of an eye containing a malignant melanoma might accelerate the dissemination of tumor cells (Fig. 11-14).
Although plaque brachytherapy conserves eyes, radiation retinopathy or papillopathy occurs commonly leading to loss of useful vision. Almost half of treated eyes have 20/200 vision 3 years after therapy. Some smaller tumors can be locally resected by partial lamellar sclerouvectomy. Today, this technique generally is performed on iridociliary tumors. Small relatively thin tumors of the posterior choroid can be treated with transpupillary thermotherapy (TTT), an infrared diode laser therapy that kills tumors cells by slowly heating them. A sandwich technique combining TTT and plaque brachytherapy has been used to prevent tumor recurrence from cells sheltered in scleral canals at the base of a tumor. Endoresection of uveal melanoma has been investigated in some centers.
If a pigmented lesion of the iris is thought to be a melanoma based on documented growth or other clinical factors, it can be locally excised by iridectomy, or by iridocyclectomy if there is focal angle and ciliary body involvement. Enucleation is often necessary when an iris melanoma has caused glaucoma or is unresectable. Enucleation also may be done after histopathologic examination has shown that a previously resected tumor is a high-grade lesion. Plaque brachytherapy occasionally is used to treat unresectable iris melanomas.
THE DIFFERENTIAL DIAGNOSIS OF UVEAL MELANOMA
The differential diagnosis of posterior uveal malignant melanoma includes other benign and malignant neoplasms such as melanocytic nevi, choroidal hemangioma, and metastases from distant nonocular primary neoplasms. The list includes other rare primary intraocular neoplasms that arise in the uveal stroma such as schwannoma, leiomyoma, hemangiopericytoma, and adenomas and adenocarcinomas of the RPE and the pigmented and nonpigmented ciliary epithelium that typically are situated on its inner surface. Nonneoplastic conditions that can simulate posterior uveal melanoma and other intraocular neoplasms include vascular and hemorrhagic lesions, Inflammatory and infectious conditions, and a variety of miscellaneous disorders. Age-related maculopathy and peripheral exudative hemorrhagic chorioretinopathy (“peripheral disciform”) are important vascular lesions. Inflammatory causes include nodular posterior scleritis, uveal effusion syndrome, and granulomas. Many of these simulating conditions are beautifully illustrated in Shields’ Atlas and Textbook of Intraocular Tumors.
Choroidal hemangiomas are benign vascular hamartomas composed of relatively large, thin-walled vascular channels lined by endothelial cells. Choroidal hemangiomas occur sporadically, or in association with the Sturge-Weber syndrome (encephalotrigeminal angiomatosis). Sporadic cases appear as discrete orange-red tumefactions (Fig. 11-18). In contrast, the hemangiomas in patients with Sturge-Weber syndrome typically are diffuse lesions that obscure normal choroidal landmarks and impart a tomato-ketchup appearance to the fundus on ophthalmoscopy (Fig. 2-12). Both types of hemangiomas frequently have an associated serous detachment of the neurosensory retina that involves the fovea. Hemangiomas can produce visual loss if they are located beneath the fovea and induce hyperopia, or if they produce a retinal detachment.
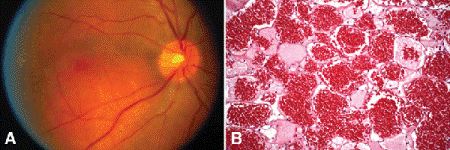
Fig. 11-18. A. Sporadic choroidal hemangioma. Discrete orange-red tumor is located beneath inferotemporal vascular arcade. The patient did not have Sturge-Weber syndrome. B. Benign vascular tumor in choroid is composed of large thin walled vessels with little intervening stroma. (B. H&E ×50)
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


