Inherited Dystrophies and Developmental Anomalies of the Cornea
Anthony J. Bron
Anthony Aldave
Saeed Akhtar
Helen S. Stewart
Ken K. Nischal
This chapter deals with inherited corneal disorders, including the corneal dystrophies and [JW1]those developmental ocular anomalies with corneal features. In recent years, with the revolution in molecular genetic research, our understanding of these disorders has changed dramatically, as disorders have been mapped and the genes responsible have been identified. It has become apparent that many disorders that were once regarded as distinct entities are examples of phenotypic heterogeneity, in which different mutations in a single gene result in a variety of clinical phenotypes. This is exemplified by the dominant corneal dystrophies-granular, lattice, and honeycomb-that are caused by separate mutations in the gene BIGH3. Similarly, mutations in PAX6, one of a family of homeobox genes controlling several transcription factors, give rise to a variety of anterior segment abnormalities including Peters anomaly, autosomal dominant keratitis, and aniridia. At the same time, examples of genotypic heterogeneity also exist, in which the same clinical phenotype results from mutations in different genes. Examples include some forms of anterior segment dysgenesis.
With these new findings and the expectation of further developments, a call has arisen for a revised classification of the corneal dystrophies, substituting a genotypic for a phenotypic approach.1,2,3,4 However, for clinical purposes, at the present time, we favor a classification based on the clinical features of these disorders, supplemented by the genetic information available. A genotypic approach, combined with knowledge of protein function, will provide increasing understanding of mechanisms and new opportunities for medical, genetic, and surgical interventions in the future.
The first part of this chapter deals with the corneal dystrophies, and the second part deals with developmental anomalies of the cornea. Some overlap occurs in these categories, because some disorders traditionally regarded as dystrophies, such as congenital hereditary endothelial dystrophy, may equally be regarded as developmental anomalies.
Loss of Corneal Transparency in the Corneal Dystrophies
Many of the corneal dystrophies give rise to a disordered physiology that is responsible for loss of corneal transparency. The normal cornea is perfectly transparent, transmitting almost 100% of visible light. Stromal transparency is achieved through the regular organization of its collagen fibrils, whose width and separation (approximately 300 nm) are less than half the wavelength of visible light. Under these conditions, forward-scattered light is cancelled out by destructive interference, and the majority of the incident light is transmitted.5 Also, although the epithelium contains organelles capable of causing significant scattering, the thinness of the epithelium in relation to the overall thickness of the cornea, and the narrow intercellular spaces, reduces scattering to an insignificant level. Similar arguments apply to the endothelium.
Two other factors that favor transparency are the avascularity of the cornea and the organization of the keratocytes. The cornea possesses properties that inhibit vascularization, such as the compactness of the nonedematous stroma. In addition it also contains the ubiquitous collagen XVIII, whose 20kDa COOH-terminal fragment (endostatin) is a potent and specific inhibitor of angiogenesis.6 The barriers to neovascularization may be lost in the presence of corneal disease. In addition, keratocytes, which have some light-scattering properties, are disposed in an antero-posterior spiral, which lessens their superimposition along the visual axis.7
The orderly packing of the stromal collagen upon which transparency relies depends in part on interactions with matrix proteins, such as the proteoglycans, and in part on the pumping action of the corneal endothelium. The endothelium possesses tight junctions that restrict the movement of solute and water. However, the permeability of the endothelium to water is about 200 times greater (in the rabbit),5 than that of the epithelium, which behaves as an almost perfect semipermeable membrane. The endothelium actively removes water from the cornea via a pump-leak system: Water is removed from the cornea at a rate that balances its leak back from the anterior chamber. The endothelium transfers ions and osmotically driven water from the stroma into the anterior chamber and creates a negative hydrostatic pressure within the stroma. This maintains the stroma in a compacted, or deturgesced state, which ensures collagen fibril order. Because no major barrier exists to the intercellular movement of water within the epithelium, the hydrostatic pressure within the stroma is transmitted to the epithelium, and the epithelium remains edema-free as long as a negative hydrostatic pressure is maintained in the stroma. This is relevant to the mechanism of corneal edema occurring in Fuchs dystrophy, discussed later in this chapter.
Disruption of the regular organization of the stroma by corneal edema, pathologic deposits, scarring, and neovascularization is responsible for the many forms of corneal opacity that characterise the corneal dystrophies.
The Corneal Dystrophies
The term corneal dystrophy is used here to define primary, inherited, bilateral disorders of the cornea affecting transparency and sometimes refraction. The form of opacity defines the clinical phenotype. Such disorders may or may not disturb vision and are generally unrelated to environmental factors. At various times in the past, the dystrophies have been said to be of early onset, axially symmetrical, slowly progressive, or stationary. Earlier definitions also required that the affected corneas were free of vascularization and unassociated with systemic disease. These are reasonable guidelines, but exceptions exist to each of them among conditions generally accepted to be corneal dystrophies. Thus, asymmetrical or even unilateral Meesmann and lattice dystrophy (LCD) occur, and vascularization is a characteristic of autosomal dominant keratitis and gelatinous drop-like corneal dystrophy. Cornea guttata and Fuchs dystrophy are late in presentation, and type II lattice corneal dystrophy is a manifestation of an inherited, systemic amyloidosis. Macular corneal dystrophy is accompanied by changes in the levels of antigenic serum keratan sulfate (KS), implying extra-ocular involvement, although in the absence of systemic disease. Corneal changes, too, are recognized features of inherited systemic diseases such as X-linked ichthyosis (steroid sulfatase deficiency),8 ceramide trihexosidase deficiency (Fabry disease),9 the mucopolysaccharidoses, and the mucolipidoses. A list of the dystrophies discussed in this chapter is given in Table 63.1 with some further details given about responsible mutations, clinical characteristics, and symptoms in Tables 63.2, 63.3 and 63.4.
Table 63.1 Clinical classification of the corneal dystrophies. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Table 63.2. Mutations responsible for the corneal dystrophies. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Table 63.3. Characteristics of the corneal dystrophies. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
Table 63.4. Symptoms in selected corneal dystrophies. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
For convenience, the corneal dystrophies are discussed topographically, starting with those disorders whose major target is the epithelium, and ending with those that chiefly affect the endothelium. But it should be recognized that the dystrophies rarely respect such precise anatomic boundaries, and some descriptive latitude must be allowed. Table 63.1 presents a phenotypic classification of the corneal dystrophies together with information about the mutations giving rise to them where this is known. Where the evidence for a specific inherited disorder has been accepted by the Human Gene Nomenclature Database Committee, the disorder receives a unique number, which is available from the website Online Mendelian Inheritance in Man (OMIM). This site is regularly updated and offers further information about the disorders. The corneal dystrophy section of the International Classification of Disease (ICD) is currently under review. Information about specific mutations is provided in Appendix 1.
The Epithelial Corneal Dystrophies
Recurrent Corneal Erosion
Recurrent corneal erosion is characterized by defective adhesion of the corneal epithelium to the Bowman layer, and by symptoms of early morning waking with pain or difficulty in opening the lids, followed by profuse watering of the eyes. The repeated attacks are due to epithelial separations. Although recurrent erosion is commonly traumatic in origin, sporadic and inherited forms also occur.
About 70% of recurrent corneal erosions are secondary to trauma, usually of a glancing nature and caused by flexible materials such as a baby’s fingernail or a twig. But the remainder are sporadic, occurring spontaneously or in response to mild trauma, and thus implying a constitutional defect of epithelial adhesion. In these cases, although a family history may be lacking, it is likely that a genetic contribution is present. A proportion of recurrent erosions are associated with map-dot-fingerprint disorder, otherwise known as epithelial basement membrane disorder (EBMD), but reports also exist of recurrent erosion inherited as a dominant trait, in the absence of clinical basement membrane signs. In a study of recurrent erosion by Reidy and colleagues, 45% were traumatic alone, 29% had EBMD alone, and 17% had a history of both trauma and EBMD.10 The total associated with trauma was 63%.
Franceschetti reported a family in which recurrent corneal erosion occurred in a dominant pedigree over six generations (Fig. 63.1).11,12 The disease started between the ages of 4 and 6 years, sometimes precipitated by trivial trauma. With increasing age, attacks were less frequent and intense, and were rare after the age of 50 years. No significant subepithelial scarring occurred, and vision remained good. Other authors have made similar reports.13,14,15,16,17,18 In the family described by Fialho Filho, involving seven cases in two generations, attacks of superficial keratitis resulted in permanent central changes.19
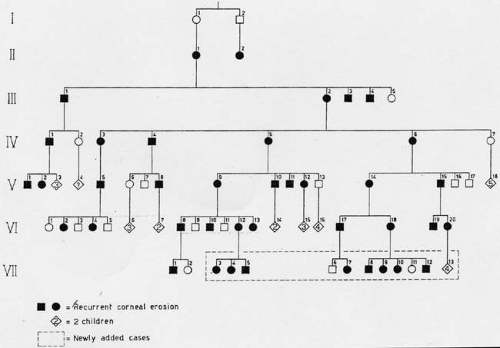 Figure 63.1. Dominant pedigree of recurrent corneal erosion. (Reproduced with permission from Waardenburg PG, Franceschetti A, Klein D, eds. Genetics and ophthalmology, Vol. 1. Oxford: Blackwell, 1961.) |
Recurrent erosive attacks may be a feature of certain inherited dystrophies of the Bowman layer or the corneal stroma, such as honeycomb or lattice corneal dystrophies, presenting in the first decade of life before the nature of the underlying dystrophy has become evident, but persisting into adult life.18 In some forms of LCD, it has been observed that severe attacks may be associated with a greater than usual degree of scarring in the more affected eye.20 This contrasts with the situation in isolated traumatic or spontaneous recurrent erosion, in which scarring is an unusual feature. Erosion attacks occurring in relation to such established dystrophies are less likely to have the distinctive features described above, so that painful episodes may occur at any time of the day or night.
A typical recurrent erosion attack is heralded by severe pain occurring in the early hours of the morning or on opening the eyes on waking. Patients may adopt manoeuvres to minimize or prevent pain on lid opening, such as light manipulations of the closed lids, or deliberate movements of the eyes prior to lid opening. The history of nocturnal or waking pain is so typical as to be pathognomonic. Pain is accompanied by marked watering, which settles as the day advances. Most attacks settle within 5 days of onset on conservative therapy.21
Recurrent erosions may take a “macro” form, with frank epithelial loss, or a “micro” form, with a lesser degree of pain and indicated by a cluster of epithelial microcysts that mark the focus of an adhesion defect (Fig. 63.2).22,23,24 That attacks of pain occur, even in the micro-form in which the epithelium is intact, suggests that pain may be a response to shearing of nerve terminals when the intact, but poorly adherent epithelium, slides transiently over the underlying basal lamina. Both trauma- and EMBD-related attacks preferentially affect the lower half of the cornea.21
 Figure 63.2. Recurrent corneal erosion: Drawing of a macro-erosion.: An epithelial detachment associated with a macroerosion. C: Drawings shows a typical cluster of epithelial microcysts. D: Post-traumatic epithelial microcysts, some containing debris. E: Epithelial microcysts staining brilliantly with fluorescein. F: Low-power TEM of epithelial microcyst in debridement specimen from a patient with nontraumatic recurrent erosion. G: High-power TEM from the same specimen showing basal epithelial edema and separation of the basal lamina. Separation from the cell may be artifactual. (Figures 2F and G, courtesy of RC Tripathi.) |
In a patient in remission but with a clinical history of recurrent corneal erosion, the demonstration of epithelial microcysts is an important and diagnostic feature of the disorder, which also provides a visible target for micropuncture therapy or phototherapeutic keratoplasty (PTK) (see below). Therefore, for diagnostic purposes, a specific search for microcysts always should be made. They are most readily detected by instilling fluorescein into the conjunctival sac, whereupon microcysts are signalled by the dark breaks that they induce in the overlying, fluorescent tear film. Occasionally they take up fluorescein and are then seen as brilliant fluorescent dots, much brighter than stained punctate erosions. Their brightness relates to the size of the microcysts, because the fluorescent luminosity depends on the thickness of the observed film. Spontaneous recurrent erosions may occur bilaterally, but when they are unilateral, the presence of epithelial microcysts in the fellow eye may confirm a constitutional disposition. A differential diagnosis of epithelial microcysts is given in Table 63.5.25,26,27,28
Table 63.5. Differential diagnosis of microcystic epithelial disease. | ||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||
Recurrent attacks cause remarkably little permanent damage to the corneal stroma, so that scarring is unusual, and patients may be reassured that vision is unlikely to be affected. However, occasionally, a severe attack leaves a corneal opacity and, very rarely, a bacterial keratitis may supervene.
An acute macroerosion is treated like a traumatic abrasion, with antibiotic ointment and mydriatic. Topical steroids may be useful in the initial stages if a marked inflammatory reaction is present. It may be necessary to débride loose sheets of epithelium. Some authors find no difference in the symptomatic recovery or resurfacing of traumatic abrasions with or without patching,29 for which reason some authors prefer to avoid the use of eye pads.30,31 Healing usually proceeds normally, because the problem is one of adhesion and no defect in epithelial resurfacing occurs in this disorder; however, recurrences are the hallmark of the condition. Following an attack, the patient must continue to use a simple lubricating ointment at night to reduce friction between the upper lid and the globe. Nightly use of hypertonic agents, such as 5% sodium chloride drops, corn syrup, or ointment has been advised, on the basis that this reduces a nocturnal epithelial edema. However, Hykin and colleagues found no difference in the efficacy of simple ointment and sodium chloride,21 and sodium chloride preparations cause significant stinging. Other conservative measures advocated in the treatment of recurrent erosion include the use of contact lenses, including gas permeable silicone hydrogel soft lenses32 and scleral lenses.33 A therapeutic hydrogel, with a relatively tight fit, is advocated, particularly for traumatic recurrent erosions in which the lesion is axial.34 In general, EMBD is a risk factor for failure of therapy, with a relative risk of over 10.21
These measures may reduce the frequency of attacks but, when they fail to afford long-term relief, a surgical approach such as corneal micropuncture or PTK may give long-lasting relief or even a permanent cure. These procedures are discussed in the section on map-dot-fingerprint disorder.
Dystrophia Helsingia
Dystrophia helsingia is a dominant form of recurrent corneal erosion, giving rise to discrete subepithelial scars in later life.35 It is inherited as an autosomal dominant trait, observed in a family of seven generations.
This is a newly described bilateral corneal dystrophy reported from Sweden in a family of six generations. Attacks of recurrent corneal erosion start at around 5 years of age in the majority of affected patients and occur about four times a year, each attack lasting about 3 to 7 days. Erosions are less frequent between the ages of 20 and 30 years, but the eyes remain sensitive to minor trauma throughout life. From the age of about 20 years, discrete subepithelial scars develop in the mid periphery and then more centrally, rarely affecting the central cornea. At around the age of 70 years, opacities are associated with small, drop-like elevations. Visual acuity is preserved.
Treatment is as for other forms of recurrent erosion.
Dystrophia Smalandia
Dystrophia smalandia is a further corneal dystrophy dominated by recurrent corneal erosions and, in this case, the development of keloid-like, axial corneal scars giving rise to visual loss in later life.35,36 It is inherited as a dominant trait.
This dystrophy has been described in Smalandia, a province of southern Sweden. The disease starts as early as 7 months of age, with frequent attacks of recurrent corneal erosion. The majority of patients are affected before the age of 5 years. The severity of attacks reduces by middle age, but about two-thirds of patients develop corneal keloids by the age of 40 to 50 years. Confocal microscopy has shown a normal epithelial architecture and thinning or absence of the Bowman layer. Visual loss has led to keratoplasty in a proportion of patients, which is followed by recurrences at the graft periphery, within a year of surgery.
Examination of keratoplasty material and a biopsy specimen confirms the presence of deficiencies of the Bowman layer, subepithelial fibrosis, and keloid formation associated with amyloid deposition.35
Vision can be restored by keratoplasty but is associated with recurrence.
Meesmann Epithelial Corneal Dystrophy
Meesmann dystrophy is a rare, juvenile, autosomal dominant epithelial disorder, characterized by scattered, bilateral, epithelial microcysts and variable symptoms of discomfort or visual loss. The dystrophy was first described by Pameijer in 193537 and more fully by Meesmann.38,39 Burns and Thiel and Behnke have provided full reviews.40,41
Meesmann dystrophy is caused by mutations in the genes (KRT3 at chromosome 12q13 and KRT12 at 17q12) encoding two cornea-specific cytokeratins, CK3 and CK12 respectively.42,43,44 Mutations in KRT12 have been reported in the highly conserved, a-helix, initiation or termination domains of the gene, which are regarded as essential for cytokeratin filament assembly.43 A mutation in KRT12 has been described in affected members of the more severe Stocker-Holt variety of Meesmann dystrophy, described in a family of over 200 members in North Carolina, twenty of whom had the dystrophy. The family were of Moravian descent and had emigrated from Saxony in 1741.2,45,46,47
Corneal changes consist of epithelial microcysts, which have been observed as early as age 7 months, associated with symptoms of irritation and photophobia. Cysts tend to increase in number with age. In its most developed form, the corneal epithelium of each eye is studded with minute, spherical microcysts, disposed with varying symmetry and sparing the perilimbal region (Fig. 63.3). Some reports describe the epithelial opacities to be in garlands and whorls in older patients.48 The clear cysts are approximately 10 to 50 (m in diameter and may contain a minute collection of debris. They may or may not stain with fluorescein. The intervening epithelium is clear or slightly hazy.25 Distribution may be random, whirled, sectoral, interpalpebral, or unilateral. Vision may be unaffected, or attacks of discomfort and visual disturbance may occur in the fourth or fifth decade.
 Figure 63.3. Meesmann dystrophy. A, B: Drawing shows the typical distribution of epithelial microcysts. C: Scattered microcysts seen on biomicroscopy in a case with low cyst density. D: Scattered microcysts in a patient with high cyst density (retroillumination against the iris). E: The same cysts seen against the red reflex (3D, E, courtesy of P Laibson). F: Light microscopic view of disordered and acanthotic corneal epithelium. Many epithelial cells contain PAS-positive fibrillar material (PAS stain, (200). (F reproduced with permission from [JW35]Irvine AD, Coleman CM, Moore JE et al: A novel mutation in KRT12 associated with Meesmann epithelial corneal dystrophy. Br J Ophthalmol 2002;86:729.) |
In the Stocker-Holt variety of this disease, which is symptomatically more severe, bilateral, interpalpebral epithelial recurrent erosions are detectable from the age of 7 months and give rise to visual impairment by the end of the first decade, together with episodic photophobia and lacrimation. Older individuals experience foreign-body sensation and a mild decrease in visual acuity.3,45,46
A few papers report the value of keratoplasty in the treatment of Meesmann dystrophy, and it may be that these few reports relate to atypical, more severe disease. Burns noted that, although the dystrophy recurred after simple debridement, it failed to do so after keratectomy or penetrating keratoplasty.40 Chiou and colleagues49 reported epithelial recurrence 6 years after a penetrating keratoplasty (performed at the age of 74 years) in a patient with sporadic Meesmann dystrophy. Microcystic changes were present on both the donor and recipient cornea. In the fellow eye, which had undergone lamellar keratoplasty when the patient was 51 years old, no microcystic recurrence was recorded at 84 years of age, despite the presence of microcysts in the host epithelium. Pathologically, in this eye, the epithelium was disorganised, and basal laminar thickening was present. These reports suggest that the epithelial manifestations of the dystrophy may be influenced by interactions between the epithelium and the corneal stroma, because otherwise it would be expected that replacement of donor epithelium by migrating host epithelium-which may take up to a year-would restore the dystrophic epithelium. Such interactions are well recognized as a part of the physiopathologic response of the cornea to injury,50,51 and the concept would benefit from further attention.
Although the appearance of Meesmann dystrophy is quite distinctive, bilateral epithelial microcystic changes also may be encountered in other disorders, including Lisch dystrophy, recurrent corneal erosion, vernal keratoconjunctivitis, meibomian gland obstruction, distichiasis, corneal insensitivity, and aqueous-deficient dry eye in young adults (see Table 63.5).25,52 The presence of lid disease or dry eye should therefore be excluded before a clinical diagnosis of Meesmann dystrophy is made and confirmed clinically by observation of microcystic changes in a parent or sibling.
Lisch corneal dystrophy also exhibits epithelial microcystic changes, but is inherited as an X-linked dominant trait. The Bowman layer an[JW2]d the stroma are normal in this condition. Ultrastructural studies have shown irregular basal laminar thickening, epithelial disorganization with frequent mitoses, and intraepithelial microcysts containing autofluorescent debris.53 Abundant glycogen is demonstrated with periodic acid-Schiff (PAS) stain in some but not all studies. The thickened basal lamina may invaginate the epithelium.40,54 The electron-dense “peculiar substance” noted by Kuwabara and Cicarelli53,55 is recognized to be the morphologic counterpart of aggregated, mutated cytokeratin (Fig. 63.3B). Such aggregates have not been identified in the Stocker-Holt disorder.3
Lisch Corneal Dystrophy
Lisch corneal dystrophy is a bilateral, X-linked dominant epithelial disorder in which epithelial microcysts are disposed in radial bands or whorls. The condition is distinct from Meesmann dystrophy.
The original pedigree56 of five family members could have been interpreted as a dominant trait,57 but further studies by the original authors, examining an expanded pedigree, have established it to be an X-linked disorder with expression in both hemizygous males and carrier females. This is an example of pseudodominance (Fig. 63.4).58 In keeping with its X-linked status, father to son transmission was not observed. Lisch dystrophy maps to the short arm of the X-chromosome (Xp22.3), and mutations in the keratin genes KRT3 and KRT12, responsible for Meesmann dystrophy have been excluded.58 Robin and colleagues reported two sporadic cases of what appears to be Lisch dystrophy.59
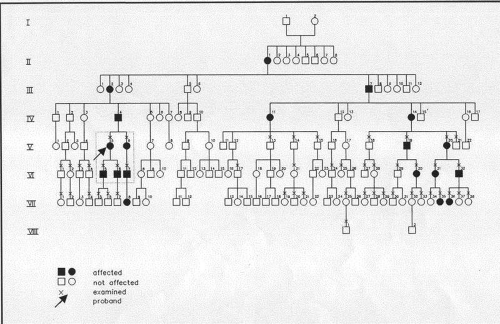 Figure 63.4. Lisch dystrophy. Pedigree showing 19 affected members in eight generations. Note the pseudo-dominant inheritance pattern of this X-linked disorder, which depends on expression of the gene in both males and females, giving rise to transmission across five generations. The absence of direct male-to-male transmission is in keeping with an X-linked trait. (Used with permission from Lisch W, Buttner A, Oeffner F et al: Lisch corneal dystrophy is genetically distinct from Meesmann corneal dystrophy and maps to xp22.3. Am J Ophthalmol 2000;130:461–468.) |
Slit-lamp examination reveals radially disposed, gray, narrow or broad, band-shaped, feathery opacities, sometimes taking on a whorled appearance and lying within larger zones of clear epithelium (Fig. 63.5). On retroillumination, they are seen to be made up of densely crowded, clear, epithelial microcysts.56 The microcysts do not stain with fluorescein. Vision may be normal, or it is reduced if the opacities lie on the visual axis. Unlike the situation in some patients with Meesmann dystrophy, the condition is not associated with pain. Debridement results in the temporary restoration of a clear epithelium, but the original features return within a few months, suggesting that they are restored as the resurfaced epithelium differentiates. Lisch and colleagues56 and possibly Robin59 noted a reduction in epithelial changes in a patient who wore hard contact lenses, implying that accelerated shedding of surface cells induced by contact lens wear removed the epithelial cells before the degenerative changes were sufficiently established to cause opacity.
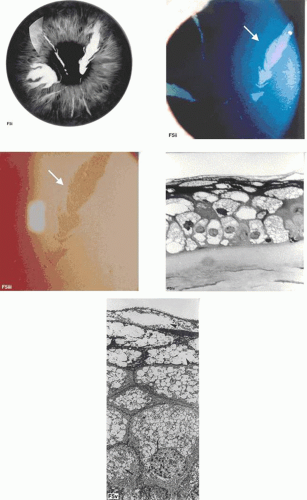 Figure 63.5. Lisch dystrophy. A: Drawing shows the radial arrangement of epithelial opacities in Lisch dystrophy, reflecting the radial architecture of the corneal epithelium. B: Slit-lamp picture of selected opacities seen in focal illumination. C: The same opacities in retroillumination. D: Light microscopy of the corneal epithelium, showing marked vacuolation of both superficial and basal epithelial cells. E: Transmission EM showing vacuolated epithelial cells in detail (arrows; N, nucleus) (B-E reproduced with permission from Lisch W, Steuhl KP, Lisch C et al: A new, band-shaped and whorled microcystic dystrophy of the corneal epithelium. Am J Ophthalmol 1992;114:35–44; photographs courtesy of W Lisch.) |
The radial disposition of some of the observed epithelial opacities in Lisch dystrophy recalls the vortex arrangement of the epithelium encountered in Fabry disease (cornea verticillata) and in some drug-induced epithelial keratopathies.60 The opacities in the latter disorders are due to the presence of multilamellar bodies within the epithelium as part of either an inherited or drug-induced lipidosis.61 These are responsible for the fine granular haze seen on biomicroscopy. The radial pattern reflects the radial architecture of the corneal epithelium, which in turn is due to the centripetal epithelial migration of epithelial cells from their limbal, stem-cell origin. The biomicroscopic opacity of the epithelium in Lisch dystrophy is due to the scattering of light by the degenerate epithelial cells. Neighboring, biomicroscopically clear regions of the epithelium show no histologic features of degeneration. In Fabry disease, scattering arises from the presence of the multilamellar bodies within the epithelial cells.
If the oblique patterns in both disorders are both due to the radial architecture of the corneal epithelium, then it is not clear why the changes affect the epithelium diffusely in Fabry disease and patchily in Lisch dystrophy. In normal XX females, half of the X-chromosomes are inactivated by the process of Lyonization,62 and it might reasonably be assumed that in a female carrier of an X-linked disorder such as Lisch dystrophy or Fabry disease, half of the limbal stem cells would produce a progeny of opaque epithelial cells and half would produce a progeny of clear cells. Consequently, it would be expected that carrier females would show a lesser degree of epithelial opacity than would affected male hemizygotes. Because the process of Lyonization is random, a relatively homogeneous mixing of cells containing one or more X-chromosomes results. This phenomenon gives rise to the peripheral retinal mottling in X-linked ocular albinism.63 As far as the authors of this chapter are aware, this dosage effect has not been reported in Lisch dystrophy. Also, Lisch and colleagues58 found no difference between the degree of opacity in males and females with Lisch dystrophy.
It is of interest that studies of the limbal stem-cell architecture in the mouse suggest that the limbus is supplied by a limited number of stem-cell clones such that small, contiguous stretches of the limbus contain stem cells arising from separate clones, with each linear block arising from a separate clone.64 Collinson and colleagues, observing chimeric animals, were able to make cells of differing genetic origin visible by labelling half the cells so that they could be imaged as blue and the rest as clear.64 This approach created an epithelial vortex pattern, the appearance of which is very like that of Lisch dystrophy (Fig. 63.6). In the Collinson experiment, the radial stripes represent groups of centripetally directed cells that are the progeny of single clones of limbal stem cells or from groups of contiguous, adjacent clones. Although it seems likely that the human limbal stem-cell system is also organized in clonal blocks, some caution must be exercised in drawing inferences, because it is difficult to explain why the radial features of the Lisch opacities are focal, while those in Fabry disease are diffuse. One possible explanation might be that cells that are the progeny of a single clone mature at the same time; then, if maturation is not synchronous between each radial clonal block, opacification occurring late in maturation occurs in sequence, now in one region, then in another. This phenomenon could explain the existence of clear and opaque regions of epithelium in Lisch dystrophy.
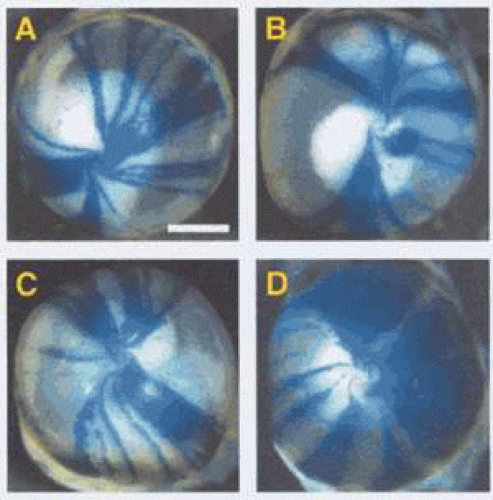 Figure 63.6. Corneas obtained from chimeric mice, 50% of whose cells carry the marker LacZ and are colored blue in this preparation. Figures A-D illustrate differing patterns of epithelial cell migration from the stem-cell source at the limbus. Each stripe represents the progeny of one or more stem-cell clones. Note that all stripes have a radial disposition, which reflects the radial organization of the corneal epithelium. This property is shared by human corneas (Courtesy of J West; reproduced with permission from Collinson JM, Morris L, Reid AI et al: Clonal analysis of patterns of growth, stem cell activity, and cell movement during the development and maintenance of the murine corneal epithelium. Dev Dyn 2002;224:432–440.) |
Light microscopy of debridement specimens showed extensive epithelial vacuolation of all cells in the affected region, particularly involving the wing cell layer, with a sharp demarcation between affected and unaffected regions (see Fig. 63.5). Electron microscopy showed incipient vacuolation in the basal layer. In one specimen, in which it was possible to examine the basal lamina, this was found to be reduplicated as in Meesmann dystrophy, but the fibrillo-granular, so-called “peculiar substance” observed in patients with Meesmann dystrophy40,53 was absent.
Epithelial Rosette Dystrophy
Epithelial rosette dystrophy is an autosomal dominant dystrophy characterized by tiny, rosette-like epithelial lesions without effect on vision. This bilateral disorder has been observed in a mother and two daughters and is characterized by tiny, sparse, elevated, focal, poorly staining epithelial lesions with a rosette-like appearance (Fig. 63.7).18 The lesions alter in position with time so that, over a period of months, they are found in any region, except the far periphery. The epithelial lesions were present from childhood and associated with mild, chronic symptoms of irritation and watering, without visual loss. Examination of the mother at the age of 62 years, 30 years after presentation, showed that small, punctate, subepithelial scars had replaced the epithelial lesions. No histopathology is available.
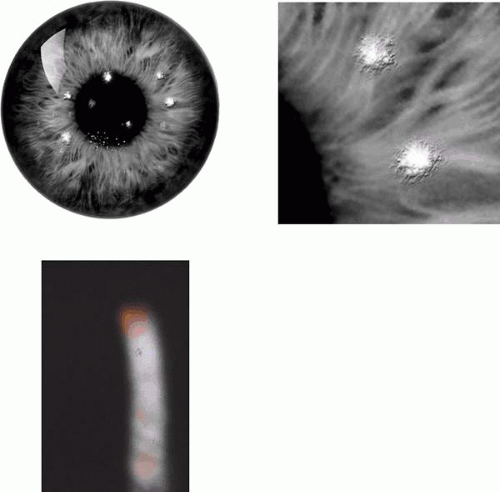 Figure 63.7. Rosette dystrophy. A: Drawing shows the sparse distribution of opacities in rosette corneal dystrophy. Inset: Detail of a subepithelial opacity. B: Slit-lamp view of a single epithelial opacity (arrow[A. J.36]). |
Map-Dot-Fingerprint Disorder or Epithelial Basement Membrane Disorder
Map-dot-fingerprint disorder is a common, mainly sporadic disorder caused by the production of a thickened abnormal epithelial basal lamina, which elevates and invaginates the corneal epithelium (Fig. 63.8). It presents after the fourth decade of life, when it may be asymptomatic or give rise to visual symptoms or recurrent corneal erosions. It is sometimes inherited as a dominant trait.65,66
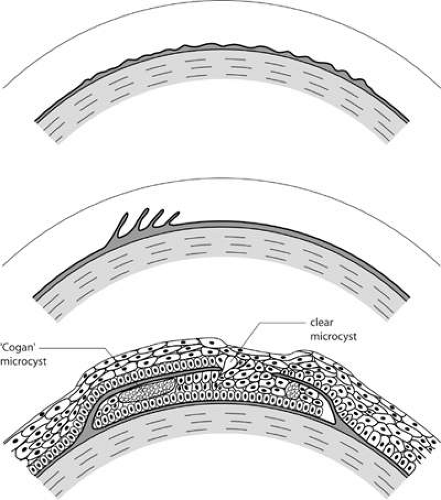 Figure 63.8. Schematic drawing to illustrate possible events in the formation of epithelial basement membrane disorders (EBMD, also map-dot-fingerprint disorder). A: A thickening of the epithelial basement membrane gives rise to the patterns of bleb and net disorder. B: A series of concentric basement membrane invaginations, roughly at right angles to the corneal surface, give rise to the appearance of fingerprint lines. C: Map changes are due to extended intraepithelial invaginations. Map regions are thickened and elevated because they give rise to a double epithelial layer. Where the invaginating layer is fenestrated epithelial cysts may gain access to the corneal surface and be shed. |
Map-dot-fingerprint disorder is a bilateral corneal disorder caused by a disturbed synthesis of basal lamina.65 Because the basal lamina is of epithelial origin, it must be regarded as an epithelial disorder. It may be entirely asymptomatic and noticed by chance during biomicroscopy, or it may give rise to visual symptoms or attacks of recurrent corneal erosion. It most commonly presents in the fourth decade of life or later. The components of map-dot- fingerprint disorder include fingerprint lines, maps and dots, bleb and net patterns23 and, rarely, mare’s tail opacities.24,67
Fingerprint lines consist of multiple, concentric, curved lines, caused by linear prolongations of basal laminar material into the corneal epithelium.68 They have a refractile appearance on retroillumination, resembling cracked glass (Fig. 63.9) and also can be detected by the breaks that they induce in the overlying, fluorescein-stained tear film. The map-like changes of map-dot-fingerprint disorder are due to an often extensive insertion of basal lamina into the corneal epithelium, parallel with the corneal surface (Fig. 63.10). This invagination splits the epithelium into two layers, causing a thickening of the epithelium in the region of the maps, a feature readily detected by fluorescein. Small fenestrations in the intraepithelial basal laminar material are associated with depressions in epithelial thickness, where the epithelium is thinned to its normal width. When maps or cysts lie on the visual axis, the disruption of the tear film may drop the acuity by a line or two.
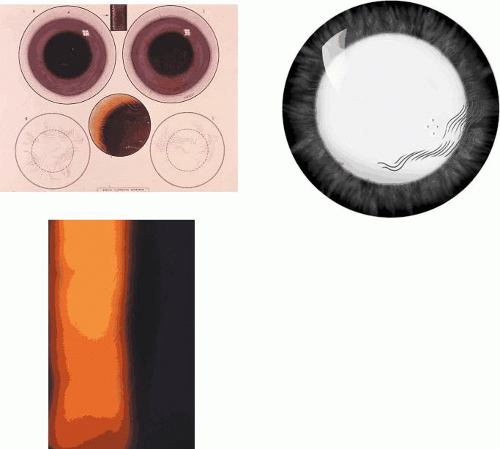 Figure 63.9. EBMD. Fingerprint lines. A: Bilateral fingerprint lines, drawn to show that the lines within each patch have a concentric arrangement. B: Drawing shows a group of fingerprint lines silhouetted against the red reflex. C: Slit-lamp view of fingerprint lines at high magnification seen by retroillumination. |
In the normal cornea, a turnover of epithelial cells occurs. Stem cells residing at the limbus divide and produce a progeny of daughter cells that migrate centripetally along the basal lamina. The further division of these “transient amplifying” cells gives rise to terminally differentiated cells, which migrate anteriorly and are shed from the corneal surface. In map-dot-fingerprint disorder, this forward migration is impeded by the intraepithelial sheets, and the migrating cells remain trapped behind the basal laminar barrier of the map lesions. Here they degenerate and form epithelial microcysts with the typical appearance first described by Cogan.69 These microcysts are responsible for the white dots of map-dot-fingerprint disorder. These large, smoothly contoured microcysts, packed with degenerate epithelial cells, are of a size, shape, and brightness of opacity quite distinct from the microcysts of recurrent corneal erosion. The latter are often clear and contain only a small amount of cellular debris. Cogan microcysts are large and opaque, and only become clear if they have the opportunity to disgorge their contents to the epithelial surface. This may occur either at the edges of an intraepithelial sheet, or otherwise at the site of a fenestration, where the invaginating basal laminar material is absent. Because these microcysts reside within a posterior epithelium that is turning over in an irregular way, they adopt a variable, smoothly plastic shape, which changes over time. Like fingerprint lines, they may affect acuity if they occur on the visual axis (see Fig. 63.10).
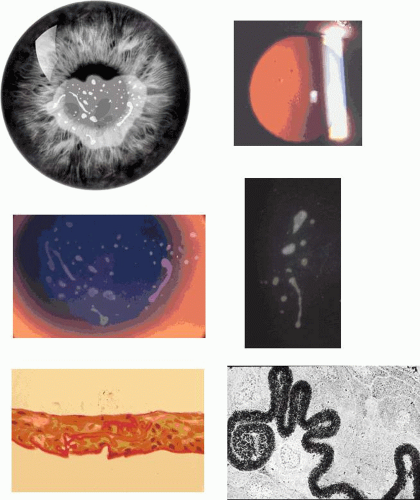 Figure 63.10. EBMD. A: Drawing of a centrally placed “map,” showing scattered, dense white Cogan microcysts, which are the “dots” of map-dot-fingerprint disorder. Note also the oval fenestrations. B: The same map is seen by retroillumination against the red reflex. The map consists of an elevated epithelial island composed of two epithelial layers separated by an invaginating basal lamina. C: High-power slit-lamp view of the map zone shown in the preceding picture viewed by oblique focal illumination. The epithelium is hazy and contains an assortment of large and small smoothly contoured microcysts, each completely filled with cellular debris. D: An isolated collection of opaque (Cogan) microcysts in another patient. E: Light micrograph of the epithelium in another patient with EBMD, showing extensive and irregular invagination of epithelium by basal laminar material (PAS stain). F: Transmission EM: High-power detail of basal laminar invagination in this disorder. Same patient as in 9 B and C (silver methenamine stain; figure F, courtesy of RC Tripathi). |
The bleb pattern resembles a sheet of pebbled glass on retroillumination23,66 and is due to the presence of subepithelial mounds of a PAS-positive material underlying a normal basal lamina.70 When complete, it takes the form of a discoid sheet, occupying up to three-quarters of the area of the cornea, or it may occur in scattered patches (Fig. 63.11). The condition is not associated with visual loss, staining, or a disturbance of the overlying tear film. The net-like pattern has the same pebbled appearance, but is arranged in a broad, net-like pattern whose dimensions correspond exactly to those of the anterior corneal mosaic, a normal architectural feature of the superficial cornea.71,72 The latter can be demonstrated readily in the normal cornea by applying firm pressure to the cornea through the closed lid and viewing in the presence of fluorescein or alternatively in the red reflex. A broad, net-like pattern becomes visible in the epithelium, which fades slowly with time.
 Figure 63.11. EBMD. Bleb and net patterns. A: Drawing of a bleb pattern seen by retroillumination, here occupying about two-thirds of the corneal surface (The size of the blebs has been slightly overexaggerated). B: High-power view of the bleb pattern, seen against the red reflex, resembles pebbled glass in appearance. It is caused by a thickening of the basal lamina, although no explanation has been found to explain the shape and size of its component parts. C: Drawing of a patch of net pattern in retroillumination. D: Slit-lamp photograph of the net pattern, against the red reflex. It is a variation of the bleb pattern in which the blebs take on elongated or spherical forms, which are arranged in a net-like formation. In this example, the net pattern is superimposed perfectly on the anterior corneal mosaic, which has been induced transiently by pressure on the cornea through the closed lids. E: The anterior corneal mosaic is a normal anatomic feature of cornea. In this picture, it is seen as a fluorescent net, again induced by pressure on the cornea through closed lids, this time with fluorescein in the tear film. F: The anterior mosaic is seen in specular reflection (courtesy of NJ Schweitzer). G: The structure responsible for the mosaic is subepithelial, but is not visible at the surface of the Bowman layer in this donor eye at normal ocular pressure (epithelium removed). The surface of the Bowman layer shows a smooth patina. H: However, when the pressure is subnormal and the cornea becomes concave, the ridges become apparent at the surface. These structures are the basis for the corneal mosaic, but their anatomy is not completely known (11G, H reproduced with permission from Bron AJ, Tripathi RC: The anterior corneal mosaic. Br J Physiol Opt 1970;25:8–13.). |
The nets of EBMD register precisely on the mosaic when the latter is induced by gentle pressure on the cornea through the lids (see Fig. 63.11).73 Like the bleb pattern, the net pattern, too, is usually present in small patches. The bleb pattern is due to the presence of a neutral mucopolysaccharide-protein complex forming a continuous layer between the basal lamina and the Bowman membrane. Hemi-desmosomes were reported to be intact by Dark,70 who suggested that recurrent erosions, when they occur, may be due to a shearing of the abnormal layer.
Mare’s tail lines resemble fingerprint lines in arrangement, but are broader and are opaque in focal illumination. Morphologically, they are horizontally disposed and draped in the manner of a handlebar moustache (Fig. 63.12). They may be found as isolated bilateral features or, less commonly, they may accompany maps, dots, and fingerprints. Like fingerprint lines, they are due to an invagination of the epithelium by basal lamina; their visibility in focal illumination may be due to the greater thickness of the epithelial invagination or to the additional presence of a subepithelial connective-tissue pannus.24,67 The horizontal disposition of the lines probably reflects the effect of the downstroke of the blink on the epithelium invaginated by basal lamina. The surface irregularity of mare’s tail lines may reduce vision if they affect the visual axis and may cause mild discomfort.
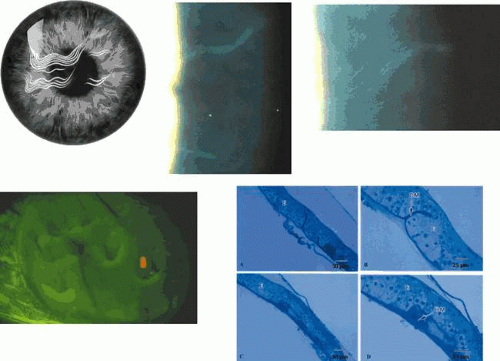 Figure 63.12. Mare’s tail lines in the right cornea of a 74-year-old woman. A: Drawing shows their disposition at the surface of the cornea[A. J.38]. B: Slit-lamp appearance of two groups of mare’s tail lines. C: Detail of the upper group shows multiple lines, concentrically arranged and disposed horizontally. D: Fluorescein staining shows that each group of mare’s tail lines is elevated and thins the overlying tear film. E: Light micrographs of débrided epithelium affected by of mare’s tail lines; A and B show invaginating folds (BM) and C and D show subepithelial accumulations of basement membrane material (semi-thin sections stained with toluidine blue stain). (Reproduced with permission from Akhtar S, Bron AJ, Meek KM: Clinical and ultrastructural findings in mare’s tail lines of the corneal epithelium. Br J Ophthalmol 2004; 88:864–867.) |
All forms of map-dot-fingerprint disorder may be associated with recurrent corneal erosion, usually presenting in the fourth decade or later, either spontaneously or in response to mild trauma. This predisposition is due to a disturbance of the normal mechanism of epithelial attachment to the Bowman layer, induced by the presence of the aberrant basal laminar material. If a macroerosion occurs, with epithelial loss, the clinical features of the disorder (maps, fingerprints, nets) on the side of the erosion may be lost with the epithelium, so that after healing only a few features of the disorder may persist at the site of the erosion, whereas they remain intact in the fellow eye. For this reason, in any patient who presents with recurrent corneal erosion, regardless of whether it appears to be traumatic, it is essential to examine both corneas after mydriasis, using focal and retroillumination and fluorescein staining. Often, the asymptomatic eye shows the features of map-dot-fingerprint disorder, whereas the eye with the erosion shows few or no changes.
Map-dot-fingerprint disorder may be an entirely asymptomatic disorder that is discovered by chance and requires no treatment. When map lesions or fingerprint lines occur on the visual axis, they may disturb vision and can be treated effectively by debridement. Recurrent erosion occurring in conjunction with map-dot-fingerprint disorder may be treated medically in the first instance, with topical antibiotics and mydriatics in the acute phase and topical lubricants at night in the chronic phase (see the section on Recurrent Erosions). However, when recurrences persist, the recommended treatment is PTK, which is also employed in the management of recalcitrant traumatic recurrent erosion. In this treatment, a superficial lamina of the Bowman layer is ablated, and the resurfacing epithelium forms a fresh basal lamina that reattaches to the subjacent stroma, laying down new attachment structures and thereby forming hemidesmosomes and integrins and reconnecting to the Bowman layer via type VII collagen (the anchoring fibrils of the epithelium) (Fig. 63.13). Initial results of PTK have been very promising, with a success rate (freedom from symptoms, or tolerable symptoms) in the region of 85%. Because map-dot-fingerprint disorder is presumed to be of epithelial origin, it must be expected that, with time, a greater risk of recurrence may be present than in traumatic recurrent erosion.
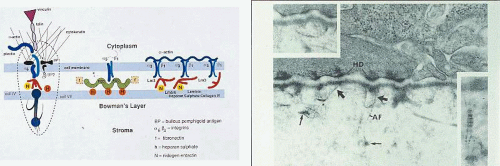 Figure 63.13. Epithelial cell attachment structures. A: Schematic diagram shows proteins involved in the attachment of the corneal epithelium to the underlying basal lamina. Epithelial cell cytoplasm and basal cell membrane (above), basal lamina (middle) and the Bowman layer and stroma (below). The figure on the left depicts the interaction between cytoplasmic proteins (actin, plectin, talin, and cytokeratin) with hemidesmosomal proteins ((6(4 integrin; bullous pemphigoid proteins [BP]), and their further interaction with laminin 5 (red figure), nidogen-entactin, and heparan sulfate in the lamina lucida, and collagen type IV (collIV) and type VII (collVII-anchoring fibrils) in the lamina densa. The collagen type VII fibrils extend into the Bowman layer. The illustrations to the right show other forms of interaction between basal cell integrins and basal lamina components. f, fibrinogen; integrins. (3(1; (5(1. B: TEM of the epithelial-basal laminar junction, showing hemidesmosomes (HD) within the plasma membrane of attached epithelial cells and anchoring fibrils (type VII) collagen embedded in the lamina densa of the basal lamin[JW39]a. (Figure 13B reproduced with permission from Tisdale AS, Spurr-Michaud SJ, Rodrigues M, et al. Development of anchoring structures of the epithelium in rabbit and human fetal corneas. Investigative Ophthalmology 1988; 29: 727–36, courtesy of I Gipson). |
Another highly effective approach for recurrent corneal erosion generally is anterior stromal micropuncture.74 Because this condition causes punctate, superficial corneal scarring, it should not be applied at or close to the visual axis. The procedure involves inducing multiple, (approximately 25 to 50) micropunctures at the level of the Bowman layer and the superficial stroma (see Fig. 63.2). Needle punctures are applied at the slit-lamp under local anaesthesia, using the presence of epithelial microcysts as a guide to the location of the treatment. Use of a bent needle tip reduces the risk of accidental corneal penetration. In the absence of such a clue to the site of the adhesion defect, it may not be possible to select a site for performing the micropuncture, and PTK may be preferable. The operation enhances adhesion by inducing scarring and a rooting down of the epithelium at multiple points around the site of defective adhesion, and by stimulating the production of extracellular matrix proteins.75,76 Postoperatively, patients are treated with a short course of antibiotics and steroid, followed by several weeks of lubricant ointment. Although the procedure itself may stimulate an erosive attack, the postoperative course is often relatively uneventful. Micropuncture is a less attractive approach for the treatment of dystrophic recurrent corneal erosion or map-dot-fingerprint disorder, because these multifocal disorders might require multiple treatments.
Recurrent corneal erosion also may be treated by simple debridement under topical anaesthesia, which is claimed to be as effective as micropuncture.77 Success is improved if debridement is carried out using a brief treatment with alcohol, in a manner similar to that used for LASIK (-alcohol delamination).78
Dystrophies of Bowman Layer and the Stroma
The Bowman layer, the anterior limiting “membrane” of the cornea, is an acellular, collagenous layer, which is destroyed in several forms of corneal dystrophy and replaced irregularly by scarring or specific deposits. The changes induced are secondary to cellular events occurring in the epithelium or subjacent stroma, so that, in this sense, no primary dystrophies of the Bowman layer exist.
Anterior Crocodile Shagreen
This consists of superficial axial, contiguous blocks of opacity of variable density, arranged in a mosaic pattern at the level of the Bowman membrane (Fig. 63.14). Dominant inheritance is reported.
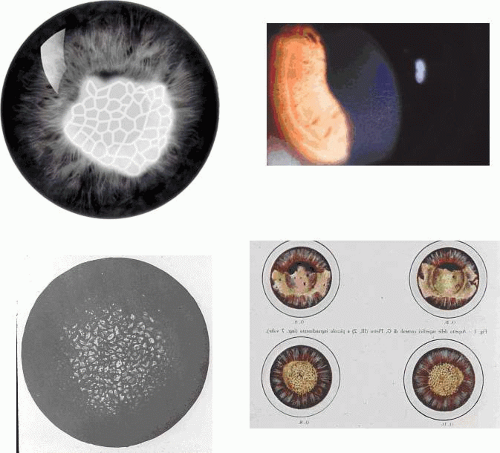 Figure 63.14. Anterior crocodile shagreen. A: Schematic drawing shows an opaque, subepithelial, mosaic opacity. B: Sporadic, unilateral, anterior crocodile shagreen in a young adult. The opacities formed a faint subepithelial network which had no effect on vision. C: An example of crocodile shagreen occurring in megalocornea. (Reproduced with permission from Boles-Carenini B[JW40]r J Ophthalmol 1961;45:64.) D: Bilateral crocodile shagreen in a son whose father had band-shaped keratopathy. (Reproduced with permission from Valerio M: Arch Ottal 1942;49:1.[JW41]) |
The condition was first described by Vogt79 in an 80-year-old man who showed flat, gray, polygonal opacities at this level. The effect on vision was small. It has been reported as a dominant disorder over two generations in a family in which the father exhibited a band keratopathy and the son exhibited anterior crocodile shagreen, calcified as in band keratopathy itself.80 Anterior crocodile shagreen has been associated with megalocornea, which is inherited as an X-linked trait.81 The net-like pattern of the disorder is not random, but, like the net pattern of EBMD, registers precisely on the anterior corneal mosaic, which resides at the Bowman level. It is assumed that in anterior crocodile shagreen, the mosaic structure determines the location of the shagreen. A peripheral, stromal shagreen is a common feature of older corneas and is not inherited (see Fig. 63.14).
Inherited Band-Shaped Keratopathy
Band-shaped keratopathy is a calcific band, at the level of the Bowman membrane, which occupies the exposed, interpalpebral zone of the cornea. It usually is due to local ocular disease and is sometimes sporadic or secondary to systemic hypercalcemia, but it may also be inherited.
Inherited band-shaped keratopathy may occur as a bilateral disorder presenting at birth, puberty, or in later life. It was found in three out of nine siblings in a family reported by Streiff and Zwahlen,82 and in two brothers, aged 66 and 71 years. The changes involved the visual axis and resembled those found in band-keratopathy secondary to local ocular disease such as uveitis, rather than the more peripheral forms associated with hypercalcemia. Calcareous band-keratopathy may occur as a secondary change in other inherited disorders such as Norrie disease83 and congenital hereditary endothelial dystrophy (CHED).84
Band-shaped keratopathy, from whatever cause, consists of a white, subepithelial opacity that lies in the exposed zone of the cornea, usually bordered by clear cornea above and below (Fig. 63.15). The band may pass from limbus to limbus, or be made up of separate central and peripheral components. Small circular holes in the opacity are typical features. The keratopathy is due to the replacement of the Bowman layer by a calcium phosphate deposit, in the form of hydroxyapatite. The deposition of this salt is explained by the ready loss of carbon dioxide from the exposed, interpalpebral zone of the cornea when the eye is open, leading to a higher pH that favors calcium deposition.
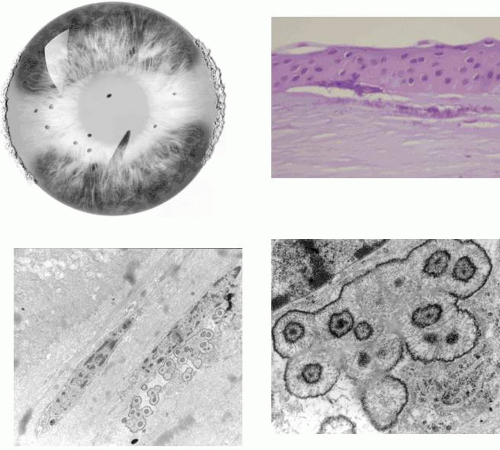 Figure 63.15. Band-shaped keratopathy. A: Drawing shows the interpalpebral disposition of band-shaped keratopathy and the characteristic scattered fenestrations within the ground-glass opacity. The vertical, white opacities at each limbus are age-related Vogt limbus girdles. B: Light micrograph of band keratopathy shows destruction of Bowman layer and deposition of calcium salts, in the presence of an avascular pannus. (H and E stain, courtesy of B MacDonald). C: Transmission EM of band keratopathy in a child with congenital hereditary corneal dystrophy (CHED). Note the absence of the Bowman layer, invasion by fibroblasts, and the presence of spherical calcium deposits, presumed to be hydroxyapatite ((5,000). D: TEM of deposits at higher power ((30,000). |
Band-shaped keratopathy affecting the visual axis causes a loss of visual acuity. Advanced keratopathy may cause symptomatic surface irregularities or painful erosions, as patches of the opacity flake off.
The opacity may be removed surgically under anaesthesia by debridement in the presence of sodium calcium edetate (Versene), which chelates the calcium and facilitates removal.
Primary Gelatinous Drop-like Dystrophy (Subepithelial Amyloidosis)
Primary gelatinous drop-like dystrophy (GDLD) or subepithelial amyloidosis presents in the first decade of life with bilateral, mulberry-like, superficial corneal elevations due to amyloid deposition. It is associated with vascularization and progresses to blindness. GDLD is an autosomal recessive disorder reported originally from Japan, but also recognized worldwide. It has recently been mapped to chromosome 1p, where mutations have been detected in the M1S1 gene, which encodes a gastrointestinal, tumour-associated antigen (Tables 63.1 and 63.2).85,86 The Gln118X mutation is most common85,87 and has been regarded as a founder mutation in the Japanese population. A Chinese patient with GDLD was found to be a compound heterozygote for mutations in the M1S1 gene, with a Gln118X mutation in one allele and a novel Y184C mutation in the other allele.88 The authors concluded that the heterozygous condition was responsible for the phenotype, but did not establish why the parent carrying the Q118X mutation did not show the disease.
Mutations in the BIGH3 gene89 and the gene for lactoferrin90a have been excluded as a basis for this disease. In a child with GDLD and global developmental delay, a mutation in the M1S1 gene was not found.90b
GDLD presents in the first or second decade with multiple, bilateral, cloudy, mulberry-like elevations of the superficial cornea. The lesions are axial and subepithelial at first, but with time invade the superficial stroma and are associated with vascularization (Fig. 63.16). It has been reported unilaterally in a patient presenting at the age of 61 years. In a study by Ide and colleagues, the same Gln118X mutation gave rise to different phenotypes (referred to as band keratopathy, stromal opacity, kumquat, and mulberry according to their appearance) in different individuals.91 The lesions stain with fluorescein due to an increase in epithelial permeability and the latter is responsible for the presence of tear lactoferrin protein within the lesions. One view of the disease is that the mutation is responsible for altered corneal epithelial permeability, which in turn leads to the diffusion of tear proteins such as lactoferrin, into the subepithelium. This is thought to lead to amyloid formation and deposition. Symptoms include photophobia, irritation, redness, and tearing, and the condition progresses to profound visual loss by the age of 20 years. Recurrence is the rule after corneal grafting.
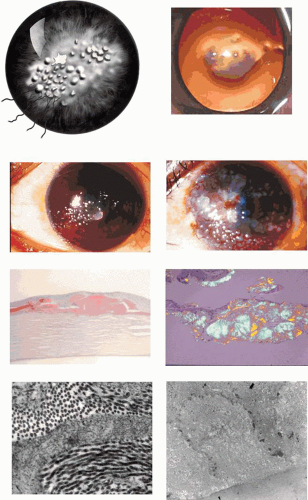 Figure 63.16. Gelatinous drop-like corneal dystrophy (GDLD). A: Schematic drawing if GDLD shows surface elevations and peripheral vascularization. B: Red reflex photograph of GDLD in a young Caucasian male, showing a gelatinous, central, superficial deposit in an avascular cornea (Reproduced with permission from Akhtar S, Bron AJ, Qin X, et al. Gelatinous drop-like corneal dystrophy in a child with developmental delay: clinicopathological features and exclusion of the M1S1 gene.Eye 2005;19:198–204.)[JW42] C: GDLD in a Japanese patient, extending to the limbus. D: A more advanced stage of GDLD in a Japanese patient, showing vascularization, hemorrhage, and a greater degree of corneal opacity. E: Low-power light micrograph showing large, irregular, subepithelial amyloid deposits in a child with GDLD (Congo red stain). F: Amyloid deposits stained with Congo red in another patient and viewed with cross-Polaroids shows birefringence (courtesy of RC Eagle). G: TEM section shows an interlamellar deposition of amyloid fibrils in the corneal stroma of the same patient (asterisks). H: TEM section shows the interepithelial deposition of amyloid fibrils in the patient shown in Fig 16B. (Figures 16C, D courtesy of Y Mashima.) |
An uncertain relationship exists between this condition and primary spheroidal degeneration of the cornea, which usually is regarded as an age-related condition. Spheroidal degeneration has, however, been described in a familial form92,93 and has been reported in combination with GDLD.94,95 The association may be fortuitous.
The drop-like elevations are due to massive subepithelial accumulations of amyloid, which progressively invade the underlying stroma. The overlying epithelium shows areas of atrophy, hyperplasia, and edema, and the Bowman layer is destroyed. With time, infiltration of the stroma is accompanied by fibrosis and vascularization (see Fig. 63.16).
Familial Corneal Scarring
Hirst and colleagues reported bilateral, localized, opaque, nodular elevations in the cornea of a patient with corneal anesthesia and dry eyes (Fig. 63.17).98 Similar lesions were found in four additional members of the family in the absence of corneal anesthesia. Histologically, the appearances were of nonspecific, corneal scarring.
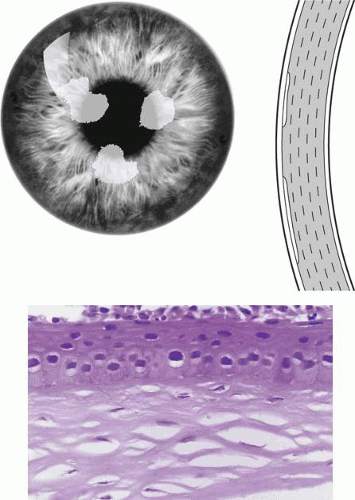 Figure 63.17. Familial corneal scarring. A: Schematic representation of familial corneal scarring shows subepithelial opacities. Sectional view (inset) shows subepithelial location of opacities. B: Light micrograph shows thinning of the Bowman layer in a region of fibrous pannus (Courtesy of WR Green). (Reproduced with permission from Hirst LW, Farmer ER, Green WR et al: Familial corneal scarring: a new dystrophy? Ophthalmology 1984;91:174–178.) |
Subepithelial Mucinous Corneal Dystrophy
Subepithelial mucinous corneal dystrophy is an autosomal dominant corneal dystrophy in which frequent recurrent corneal erosions, occurring in the first decade, are followed by decreased vision in adolescence.99
Biomicroscopy shows a bilateral, subepithelial haze, densest axially but affecting the entire extent of the cornea (Fig. 63.18). Structural studies demonstrated a subepithelial band of eosinophilic material staining positively with PAS reagent, which was Alcian blue positive and hyaluronidase sensitive. Immunohistochemical studies show the presence of a combination of chondroitin-4 sulfate and dermatan sulfate in the deposits. The condition is said to resemble Grayson-Wilbrandt dystrophy, which is a less symptomatic form of honeycomb dystrophy.
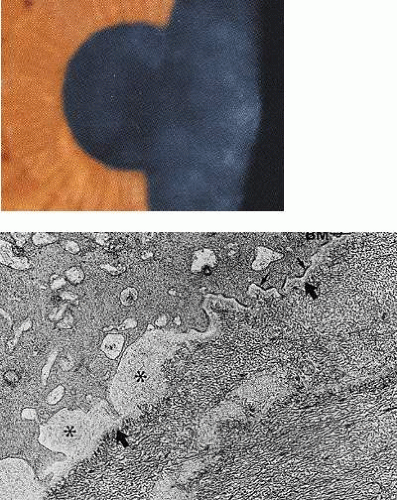 Figure 63.18. Mucinous corneal dystrophy. A: Diffuse subepithelial corneal opacities in a patient with mucinous corneal dystrophy. B: TEM of the epithelial subepithelial junction showing normal attachment structures (arrows) and lamina densa (heavy arrows). Hemidesmosomes are absent at the site of subepithelial deposits (asterisks). (Reproduced with permission from Feder RS, Jay M, Yue BY et al: Subepithelial mucinous corneal dystrophy. Clinical and pathological correlations. Arch Ophthalmol 1993;111:1106–1114.) |
The Classical Stromal Dystrophies
As their molecular basis has been unraveled, three phenotypically distinct, dominant stromal dystrophies and some of their variants have attracted considerable attention over the last two decades. They have been shown to be caused by a variety of mutations in a single gene, BIGH3 (β-transforming growth factor Induced Gene in Human clone 3). An alternative terminology used by the human gene nomenclature committee is TGFBI (TGF β-Induced gene) and TGFBI protein, for its product.3 TGFBI protein also has been called keratoepithelin, although the protein is not specific to epithelium and is found in serum and in other tissues in the body.3 These terms will be used interchangeably in this chapter.
Between 1994 and 1996, three reports appeared linking granular,100 lattice,101 and what was then termed “Reis-Bücklers” dystrophy102 (now correctly referred to as honeycomb dystrophy) to chromosome 5q31. Another dystrophy combining the biomicroscopic features of granular and LCD (“granular-lattice” or Avellino dystrophy) had been described shortly before this,103,104,105,106 mapping to the same locus.107 It was mooted that these disorders, hitherto regarded as discrete clinical entities, might be allelic. Korvatska and colleagues refined the locus of an assumed allelic gene to a 1cM region of 5q (5q22-5q32) and excluded SPARC and LOX as candidate genes.108 Later, the same group reported missense mutations in BIGH3, a candidate gene located at this locus, in family members of each of the dystrophies mapping to 5q.109,110 Mutations at codon 124 were associated with lattice dystrophy (Arg124Cys) and Avellino dystrophy (Arg124His), whereas mutations at codon 555 were associated with granular dystrophy type I (Arg555Trp) and Reis-Bücklers dystrophy (Arg555Gln). These studies have been confirmed and extended by others, and it is apparent that mutation hot spots occur at codons 124 and 555, but that mutations involving other codons also are associated with dystrophies of the same clinical phenotype. These include mutations Arg124Leu (in GCD III, a superficial form of granular dystrophy.111,112 Leu527Arg (in type IV LCD113), and Pro501Thr (LCD IIIA114) (see Table 63.1 and Appendix 1).
The gene BIGH3 was first identified in cultured adenocarcinoma cells by Skonier,115 and its product was subsequently given the name keratoepithelin,110,116 because it is strongly expressed by the corneal epithelium; it contains a signal sequence that is necessary for secretion. Keratoepithelin is expressed to a lesser extent by other corneal cells. In the normal stroma, the strongest immunoreactivity for keratoepithelin is in the Bowman layer, to a lesser extent at interlamellar locations, and to a minor degree at the interface between stroma and the Descemet layer.117
The factors leading to the particular morphologies of these dystrophies are still under debate, but most evidence supports the view that the stromal disorder results from the diffusion of a mutated protein from the corneal epithelium into the stroma where it aggregates and forms deposits. The deposits disrupt stromal organization and give rise to the typical clinical opacities. Interestingly, these dystrophies share a single negative feature in that the most peripheral 1- to 2-mm rim of perilimbal stroma remains clear and unaffected in each disorder.
Keratoepithelin is a 68 kD, 683-amino acid protein (about the size of albumin).118 Like albumin, it is sufficiently small to diffuse across the stroma from an epithelial source,119 which may explain how it comes to be bound covalently to stromal type VI collagen in normal stroma. The diffusibility of mutated keratoepithelins could explain the slow anterior-to-posterior evolution of granular and lattice dystrophies over time. Although it is not excluded that keratocytes and endothelial cells may contribute to the formation of deposits in the keratoepithelin or BIGH3 dystrophies, one of the strongest pieces of evidence supporting a major epithelial origin is the initial confinement of recurrences following keratoplasty or PTK to the epithelium and subepithelial zone (Fig. 63.19).120 This has been confirmed ultrastructurally.121 With time, recurrences spread to affect the stroma.122 However, recurrence of granular dystrophy has been reported at the graft interface following lamellar keratoplasty123 and, in another report, an increase in granular deposits appeared at the interface of flap and stroma, 16 months after bilateral LASIK surgery in a patient with granular dystrophy type 2 (Avellino).124 It is likely that, in these cases, the fresh deposits were generated by interfacial stromal fibroblasts. Although keratoepithelin is expressed in the eye primarily by corneal epithelial cells, it also is expressed by stromal fibroblasts and endothelial cells in healing corneal wounds,125 and it is likely that the intrastromal deposits in these reports are a stromal healing stimulated by the release of TGF-β.
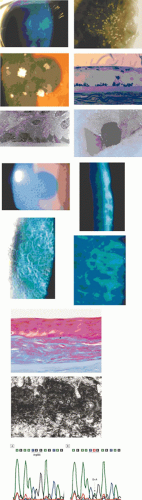 Figure 63.19. A: Recurrence of TGFBI corneal dystrophies following corneal transplantation. i. Slit-lamp pictures shows recurrences of (i.) Thiel-Behnke corneal dystrophy and (ii.) granular dystrophy (GCD I) (courtesy of RL Abbott); (iii) recurrence of Avellino dystrophy (GCD II) following PTK. Note the superficial disposition of the recurrences in each case. Light microscopic picture (iv) shows a recurrence of granular corneal dystrophy in a corneal graft. Note the dark-staining, angular subepithelial deposits and the intact Bowman membrane (semi-thin sections stained with toluidine blue). [JW43] (v-vi) TEM pictures of deposits in the same patient. B: Thiel-Behnke dystrophy. (i) Slit-lamp picture showing the typical “vitreous enamel” appearance of the corneal surface (courtesy of RC Tripathi; case of GW Cibis). (ii) Slit image of the superficial cornea in another patient. (iii) oblique slit-image to show the vitreus enamel appearance (iv) high-power slit-lamp view of the same cornea. (v) LM section of cornea stained with Masson trichrome, shows deposits that have replaced Bowman membrane ((250) (courtesy of RC Eagle). (vi) TEM shows a subepithelial deposit of “curly filaments” that have replaced the Bowman layer and project into the epithelium. Direct sequencing of exon 12 of the TGFBI gene (vii) shows: A: Normal sequence of nucleotides at codon 555 (CGG) encoding for arginine. B: A heterozygous missense mutation within this codon (at N) in an affected patient gives rise to the sequence CAG leading to the replacement of an arginine residue by the amino acid glutamine (i.e., Arg555Glu). (Reproduced with permission from [JW44]Kobayashi A, Fujiki K, Murakami A, et al: In vivo confocal microscopy and genotyping of a family with Thiel-Behnke (honeycomb) corneal dystrophy. Arch Ophthalmol 2003;121:1498–1499.) |
It was noted earlier that the keratoepithelin dystrophies have in common a clear zone of unaffected, peripheral corneal stroma about a millimeter wide. The basis for this is not known, but may reflect either removal of the diffusing, mutated keratoepithelin by the limbal blood vessels, or alternatively, that the epithelial expression of keratoepithelin is lower in the periphery, where the epithelial mitotic rate is high and epithelial cells are less mature than in the central cornea.126,127 This deserves further study.
Other factors may influence the further evolution of these dystrophies. In granular dystrophy, it has been shown that stromal deposits are aggregates of a number of proteins, including keratoepithelin itself117 and proteins such as vimentin (an intermediate filament protein),128 cytokeratin 18 (an epithelial protein),128 immunoglobulin IgG and (light chains,129 and microfibrillar protein.130 The inference is that mutated keratoepithelins, diffusing predominantly from the epithelium, coaggregate with an assortment of other proteins over a long period to form the deposits characteristic of the disorder. The activation or aggregation process also may involve some environmental influences, as suggested by a report in which recurrence of granular dystrophy in a patient who had been grafted bilaterally only occurred in the cornea that was not subjected to postoperative contact lens wear.131 Also, it has been observed that dystrophic opacities in one form of lattice corneal dystrophy were accentuated by erosive episodes, suggesting that cytokines involved in the inflammatory response (e.g., TGF-() may activate the BIGH3 gene.20
Lattice dystrophy type I is characterized by the formation of intrastromal amyloid deposits. Some, but not all, the mutations occurring at the codon 124 hot spot appear to induce structural changes that favor amyloid formation, but mutations at more than 12 other sites also have been responsible (Appendix 1). Amyloid itself is not a single chemical entity, and it is encountered in a wide variety of chronic disorders. At least 16 chemically different forms of amyloid exist, sharing a tertiary structure that is rich in (-pleated sheets. This feature is responsible for its staining properties and for the typical birefringence in polarized light. It appears that mutations leading to the keratoepithelin lattice dystrophies favor the formation of keratoepithelin-amyloid. However, other materials also are found within aggregates, including amyloid P protein132 and native gelsolin.133 In granular-lattice dystrophy, granular and lattice lesions occur in combination, with the granular lesions appearing earlier than the lattice lesions. This is a reminder that earlier histologic accounts of granular dystrophy drew attention to the occasional presence of amyloid among the deposits.134
Munier and colleagues reviewed the spectrum of mutations in 61 families exhibiting clinical phenotypes typical of the TGFBI dystrophies, using a combined approach of single-stranded conformational polymorphism (SSCP) and direct sequencing of both strands of all exons of BIGH3.135 Mutations were identified in about 80% of affected patients. A study of these mutations and of the genotype-phenotype correlations allowed two groups to be identified:
Group A patients were confined to Arg124 and Arg555 mutations, which specified the Thiel-Behnke (Arg555Gln); GCD I (Arg555Trp and Arg124Ser); GCD II (Avellino), (Cys124His), GCDIII (Reis-Bücklers or Arg124Leu); and LCD I (Arg124Cys) phenotypes. One additional, complex mutation occurred: (Arg124Leu with Thr125-Glu126) in a form of GCD I. In several families with the GCD II and GCD III phenotypes, no mutation was identified in the reading frame. Mutations were identified in 83% of this group.
Mutations at other than the 124 location reside in or at the boundary of the fasc4 domain of the gene, extending between residues 502 and 632, inclusive. The fasc 4 domain is one of four domains showing homology with the fasciculin 1 gene of drosophila. Fasciculin 1 is involved with cell differentiation and is expressed in growing nerve cones, where it is involved in the guidance of cone growth.136,137 Munier and colleagues suggested that mutations affecting the fasc 4 domain could influence corneal modelling during development.135
Group B patients exhibit a continuum of lattice phenotypes excluding LCD 1 but including LCD IIIA, LCD I/IIIA, and deep LCD. Ten different mutations, found in 79% of this group, accounted for these phenotypes. These are discussed further in the section on BIGH3-associated dystrophies.
In summary, these disorders are thought to be due to the diffusion of mutated keratoepithelin proteins from the corneal epithelium, where they chiefly originate, and into and across the Bowman layer and stroma, where they aggregate or coaggregate to form amyloid or other deposits. As will be noted shortly, in the case of honeycomb dystrophy, which is confined to the Bowman layer, it must be assumed that the aggregate or the mutated keratoepithelin itself has either a high affinity for the Bowman layer, or that its diffusion is limited by steric and size constraints or other interactions. It may be relevant that immunolocalization of native keratoepithelin is strongest at the level of the Bowman layer in the normal cornea.117
Honeycomb Corneal Dystrophy
Honeycomb corneal dystrophy (CDBII) (Thiel-Behnke) is a dominant dystrophy causing a honeycomb-like opacification of the Bowman membrane, painful corneal erosions from childhood, and progressive visual loss.138,139 (It also has been referred to as Bowman layer dystrophy, CDB type II, whereas true Reis-Bücklers dystrophy-granular dystrophy type III-is designated CDB I) (Table 63.6).140,141
Table 63.6. Dystrophies of the Bowman layer. | ||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
Okada and colleagues142 reported a BIGH3 mutation (Arg555Glu) in a patient with honeycomb dystrophy, and this dystrophy may therefore be accepted as one of the keratoepithelin dystrophies. In one report of a family with clinical honeycomb dystrophy, affected members mapped to chromosome 10q23-24, which does not carry the keratoepithelin gene.143 Thus, it appears that mutations in at least two separate genes can give rise to the Thiel-Behnke, honeycomb phenotype, an example of genetic heterogeneity in the corneal dystrophies. For reasons cited in the next section, it is likely that most of those cases referred to as Reis-Bücklers dystrophy in the past will be reassigned to Thiel-Behnke dystrophy.
Thiel and Behnke144 described an autosomal dominant dystrophy affecting the Bowman layer in 55 members of a family, traced over 11 generations. The dystrophy presented in childhood with infrequent recurrent erosions in early life, which gradually subsided in the fourth decade. A characteristic, bilateral, honeycomb arrangement of subepithelial corneal opacities appeared between the ages of 10 and 20 years, affecting all but a peripheral 1- to 2-mm of the cornea (see Fig. 63.19). The opacities increased with time, but did not affect stroma deep to the Bowman layer. Generally, the corneal surface was smooth, sensation was normal, and vision was reduced by only a moderate amount. Corneal grafting was nonetheless indicated in some patients.
The dystrophy described at that time was not “new” in the sense that it had never been seen before, but other observers had mislabelled the condition as Reis-Bücklers dystrophy, and this mislabelling has continued up to the present time.145,146,147,148,149 Confusion arose between this condition and the superficial corneal dystrophy reported by Reis150 and Bücklers.151 The latter condition was described as a disorder presenting in childhood and associated with frequent recurrent erosion attacks, progressive subepithelial opacification and severe visual loss. Weidle138,139,152,153 and others154 have pointed out that the condition described by Reis and Bücklers is a superficial form of granular dystrophy affecting the Bowman layer. This is now referred to as “geographic granular dystrophy”(granular dystrophy type III), a descriptive label that distinguishes it from the honeycomb disorder. Like other forms of granular dystrophy, it is characterized ultrastructurally by the presence of rod-shaped deposits in the corneal stroma.140,155 Other reports of a superficial form of granular dystrophy may correspond to this entity.156,157 The honeycomb dystrophy of Thiel and Behnke has a distinctly different appearance by electron microscopy, being characterized by the presence of electron-dense, curly filaments.140,141,147
Another corneal dystrophy, resembling Thiel-Behnke dystrophy in appearance, is Grayson-Wilbrandt dystrophy. This is an autosomal dominant, anterior limiting membrane dystrophy, with an onset at the end of the first decade. Unlike the situation in honeycomb dystrophy, recurrent erosion symptoms are mild.158 The extent of subepithelial opacity is variable in different family members, and distribution over the cornea may be patchy. Vision ranges from 20/20 to 20/200. Genetic studies have not yet been reported.
As noted, the ultrastructural hallmark of honeycomb dystrophy is the presence of masses of electron-dense, curly filaments that replace the Bowman membrane and the contiguous stroma and invaginate the overlying epithelium to produce surface irregularities (see Fig. 63.19).140 The connective-tissue scar contains laminin and bullous pemphigoid antigen, supporting an epithelial origin for the deposit.159
Granular Corneal Dystrophy
Granular dystrophy is a dominant dystrophy that exists in three chief forms. The classical form causes multiple, discrete, crumb-like stromal opacities; a superficial, confluent form of early onset also exists. An additional variety (granular-lattice or Avellino dystrophy) exhibits crumb-like opacities in combination with lattice-like features of later onset.
The granular dystrophies are due to mutations in the BIGH3 gene (see Table 63.1 and Appendix 1). Penetrance, as in the other BIGH3 dystrophies, is 100%.160
Klintworth2 uses the abbreviation GCD I to describe the classical stromal form of granular dystrophy, GCD II to describe granular-lattice or Avellino dystrophy, and GCD III to describe “true” Reis-Bücklers dystrophy (also referred to as Bowman dystrophy type I or CDB I140 and geographical dystrophy).
GCD I is characterized by bilateral, symmetrically grouped, bread crumb-like stromal opacities separated from one another and from the limbus by clear stroma (Fig. 63.20). Initially, subepithelial lesions are flattened and placoid in appearance, but opacities increase in number and size with age and are found increasingly in the deeper, but rarely the deepest stroma.160 The peripheral 2 mm or so of the stroma remains clear. It presents in childhood with erosive attacks and is slowly progressive, leading to significant visual loss (20/40 or worse) after the age of 40 years, which may necessitate keratoplasty. Presentation may be as early as 3 years of age.161 This variety made up 60% of the series reported by Weidle.152,153 Generally, GCD I is a less disabling disorder visually and symptomatically than is lattice dystrophy (LCD I). GCD I usually is associated with an Arg555Trp mutation in the BIGH3 gene, and is rare in Japan.162 Another mutation, Arg124Ser, described in a single Asian patient, gives rise to the same clinical picture. It is of interest that other missense mutations at the same codon (Arg124Cys and Arg124His) give rise to LCD I and GCD II (Avellino dystrophy), respectively, and to the expression of amyloid within corneal deposits.
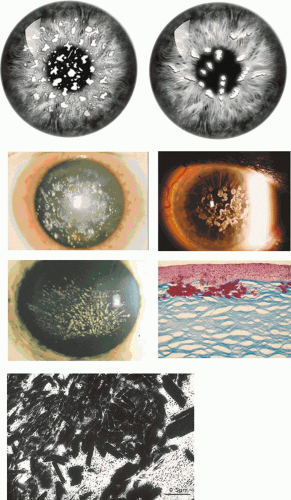 Figure 63.20. Granular corneal dystrophy. A, B: Drawing shows two forms of GCD I, one with granular opacities that are randomly scattered through the stroma, and the other with opacities that have a radial distribution. C-E: Slit-lamp pictures show different clinical forms (C) fluffy bread crumb-like stromal deposits D: Discrete and confluent ring-shaped opacities (courtesy of RL Abbott). E: A radiating pattern of deposits, reflecting the relationship between the superficial stromal deposits and the radial architecture of the corneal epithelium. Note that, in each patient, the peripheral cornea is free of changes. F: LM section of cornea, stained with Masson trichrome, showing typical red-staining subepithelial deposits with a mild disturbance of the overlying epithelium (courtesy B McDonald). G: TEM shows characteristic rod-shaped stromal deposits (courtesy of RC Tripathi). |
GCD II, granular-lattice corneal dystrophy, or Avellino dystrophy expresses the clinical and histologic features of both granular and lattice dystrophies. The original families were traced to the Campania region of Northern Italy, which includes the town of Avellino, after which the dystrophy was first named.105,163 However, since these early studies, the dystrophy has been reported from many countries, and it is the most common form of GCD in Japan162,164,165,166 and Korea.167 Thus, the name “Avellino dystrophy” is used less frequently now. GCD II corresponds to the type II granular dystrophy described by Weidle.152,153 The dystrophy is caused by an Arg124His mutation in the BIGH3 gene.110,168
It presents in the first and second decade of life with flat, subepithelial, or crumb-like stromal lesions taking on ring-shaped, discoid, star-shaped or snowflake forms (Fig. 63.21). Lattice lines appear in the stroma in later life. The proportions of each type of lesion vary widely within a sibship. Differential diagnosis from GCD I may be difficult in the absence of histologic or mutational evidence and, even here, amyloid deposits are often insignificant.3 The Arg124 His mutation in TGFI is the most common. The disorder is milder than both lattice dystrophy type I and GCD I. Progression is slower than in GCD I, and vision may only be mildly affected. Recurrent erosions, which affect women more frequently than they do men, occur less often than in GCD I (14% compared with 61% in GCD I). In the later stages, vision varies between 20/20 and 20/400 and may sometimes warrant keratoplasty.
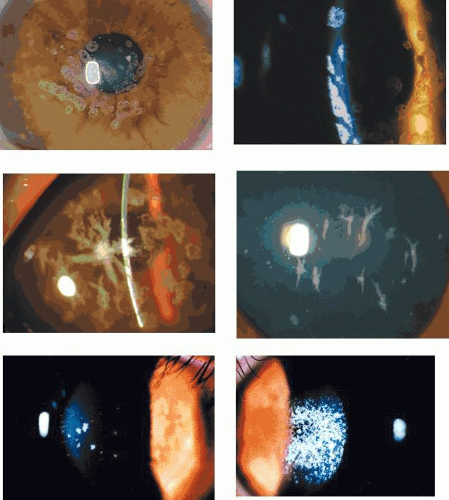 Figure 63.21. Avellino corneal dystrophy (GCD II). A-D: Differing phenotypes. A: Flat, round subepithelial opacities with a slightly radial arrangement. B: The same opacities seen in focal and retroillumination. C: Coarse, star-shaped, and irregular discoid figures are present in the superficial stroma of another patient. D: More delicate star-shaped figures are seen in a further patient. E: Early Avellino dystrophy in a 27-year-old woman indicated by a few star-shaped stromal opacities. F: In the fellow eye of the same patient, treated with LASIK 5 years previously, a massive accumulation of deposits is present at the interface of the flap and the posterior stromal bed. (Reproduced with permission from Jun RM, Tchah H, Kim TI et al: Avellino corneal dystrophy after LASIK. Ophthalmology 2004;111:463–468; pictures courtesy of RM Jun.) |
Severe, early, superficial forms of granular dystrophy have been described as homozygous manifestations in the offspring of consanguineous marriages in which both affected parents suffered from a milder disease.169,170,171 The homozygous form of GCD II is also a severe disorder, presenting by the age of 6 years and leading to keratoplasty in the first or second decade.111 The homozygous form is associated with early recurrence after surgery.111 The absence of nonocular disease in these affected children reinforces the view that keratoepithelin does not play a critical role in noncorneal tissues (Fig. 63.22).
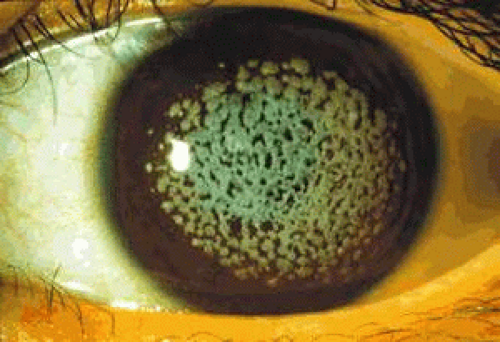 Figure 63.22. Confluent central opacities in a patient with the homozygous form of Avellino dystrophy (GCD II). (Reproduced with permission from Mashima Y, Konishi M, Nakamura Y et al: Severe form of juvenile corneal stromal dystrophy with homozygous R124H mutation in the keratoepithelin gene in five Japanese patients. Br J Ophthalmol 1998;82:1280–1284; courtesy of Y Mashima.) |
GCD III (true Reis-Bücklers’ dystrophy) is the severe, superficial variant of granular dystrophy, originally described by Reis150 and Bücklers (Fig. 63.23).151 It presents in infancy with painful erosions and is associated with fine granular, subepithelial opacities that spread and become confluent with time.154 [JW4][Wittbol-Post 1989]. This “geographic” morphology distinguishes it clinically from the “honeycomb” morphology of the Thiel-Behnke disorder.
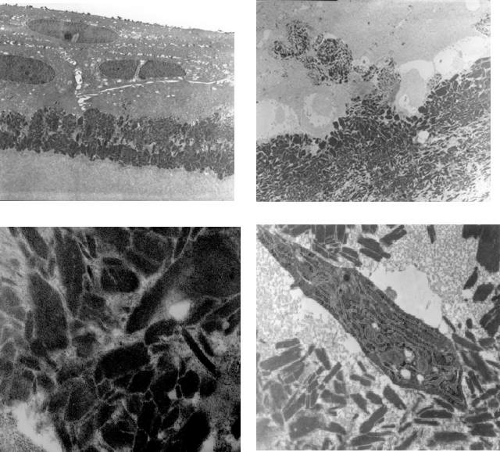 Figure 63.23. Granular dystrophy type III (true Reis-Bückler’s dystrophy; also, corneal dystrophy of the Bowman layer, type I-CDB I). TEM pictures of GCD III. A: Typical rod-shaped subepithelial deposits have destroyed the Bowman layer. Note the widened intercellular spaces. B: Fine rod-shaped deposits extend forwards between the basal epithelial cells, which are separated from their basal lamina. C: Massive intraepithelial rod-shaped deposits, some exhibiting an unusual banding pattern with a periodicity of about 10 nm. D: Rod-shaped stromal deposits in the vicinity of an activated fibroblast (Related material presented in Ridgway AE, Akhtar S, Munier FL et al: Ultrastructural and molecular analysis of Bowman’s layer corneal dystrophies: an epithelial origin? Invest Ophthalmol Vis Sci 2000;41:3286–3292.) |
GCD IV, a French variant of GCD, was described by Dighiero and colleagues as having clinical features between those of GCD I and III. Affected corneas show round, snowflake opacities in the subepithelial and most anterior stroma, with the onset of recurrent erosions in early childhood.172,173 Affected patients showed a compound heterozygous mutation, including the familiar Arg124Leu mutation associated with GCD III, and a novel mutation predicting the deletion of two amino acid residues-threonine and glutamic acid-at codons 125 and 126.
The pathognomonic ultrastructural feature of all forms of granular dystrophy is the presence of pseudocrystalline, rod-shaped deposits within the stroma (see Fig. 63.23).174 Similar rod-shaped aggregates are found within epithelial recurrences (see Fig. 63.23).
Lattice Corneal Dystrophies
The LCDs derive their name from the presence of branching, opaque or refractile, amyloid-containing filaments in the corneal stroma. The conditions are usually bilateral in presentation, but occasionally may be unilateral.175 The majority, but not all forms, are autosomal dominant disorders due to mutations in BIGH3. LCD III is a recessive disorder that has not been mapped. LCD type II is a recessive disorder due to mutations in the gelsolin gene, and is associated with a systemic neuropathy (see Table 63.1).
The BIGH3-related Lattice Dystrophies
Lattice Dystrophy type I, III, IIIa, IV-VII
LCD I is the most common form of lattice dystrophy, inherited as an autosomal dominant trait and presenting at the end of the first decade with erosive symptoms and visual difficulty. Rarely, it has been reported by 2 years of age.176 Delicate, branching, lattice-like lines occur anteriorly and axially and spread posteriorly and centrifugally with time, leaving the peripheral 1 mm or so clear (Fig. 63.24). The Descemet membrane and the endothelium are not affected. Erosive attacks usually begin in the first decade of life and are associated with increasing central stromal clouding (see Fig. 63.24), visual loss and loss of corneal sensation. In some family members they may precede the onset of characteristic stromal lesions.177,178,179 Lattice-like lines are not always readily identified,180 and they may be absent in some family members,181,182,183 thus emphasizing the value of molecular genetic diagnosis in such cases. Corneal changes may be asymmetrical, and the condition is sometimes unilateral. The clinical course varies, both within and between families, but progression usually leads to substantial discomfort and visual loss by the sixth decade. Keratoplasty is frequently indicated by the fourth decade and sometimes earlier. In two sisters grafted for LCD I, Brodovsky and Taylor184 reported the occurrence of macroerosions on the grafts, in the absence of clinical recurrence of the dystrophy, thus implying an adhesion defect expressed by resurfacing host epithelium and reinforcing the concept of a major contribution of epithelium to the disease process. LCD1 may be caused by mutations at codon 124 and at several other sites within the BIGH3 gene (see Table 63.1 and Appendix 1). El-Ashry reported intrafamilial phenotypic heterogeneity associated with the Arg124Cys mutation in Bangladeshi families.185
 Figure 63.24. Lattice dystrophy type I. A: Drawing shows lattice-like lines in the central stroma with a clear zone in the periphery. B: Low-power slit-lamp view shows branching stromal opacities by retroillumination and a central stromal haze. C: High-power view of branching lattice-like lines in another patient. D: Advanced lattice dystrophy associated with diffuse central stromal clouding. E: Low-power light micrograph showing fusiform and linear deposits of amyloid in the anterior and mid-stroma. Note the associated subepithelial scarring with destruction of the Bowman membrane (H and E stain). F: LM view shows a marked subepithelial accumulation of amyloid in the cornea of another patient (Congo red stain; courtesy of B McDonald). G: LM section of LCD I shows birefringence of a stromal amyloid deposit (Congo red-stain, viewed with crossed Polaroids). H: TEM of amyloid fibrils within a deposit, in close relation to a stromal keratocyte (courtesy of RC Tripathi). I: TEM. Amyloid fibrils located between stromal lamellae in another example of LCD I. J-L: Lattice dystrophy type IIIA. (J) Scleral scatter photograph shows ropy, thick lattice-like lines of LCD IIIA; (K) multiple branching lattice-like lines seen in retroillumination, in another patient; (L) amyloid deposits located particularly in the mid stroma and deep to the Bowman layer in LCD IIIA (Congo red stain; viewed in polarized light). (Reproduced with permission from Stock EL, Feder RS, O’Grady RB et al:Lattice corneal dystrophy type IIIA. Clinical and histopathologic correlations. Arch Ophthalmol 1991;109:354–358; photographs courtesy of EL Stock.). M-Q: Polymorphic amyloid dystrophy: (M) Representative slit-lamp pictures of polymorphic amyloid dystrophy observed against the red reflex. (N) Note the presence of branching, linear opacities in the corneal stroma (O) note the “chipped-ice” opacities in (P). A: Light micrograph of the co[JW45]rnea from a 49-year-old man with a 19-year history of symptoms, showing the wide distribution of deposits, more marked in the posterior stroma (Congo red stain). B: Birefringence under polarized light, shows the deposits to be amyloid. (Reproduced with permission from Eifrig DE, Jr., Afshari NA, Buchanan HWT et al: Polymorphic corneal amyloidosis: a disorder due to a novel mutation in the transforming growth factor beta-induced [BIGH3] gene. Ophthalmology 2004;111:1108–1114.) |
Lattice type III was first described in a Japanese pedigree and is regarded as an autosomal recessive disorder. It has not been encountered in more than one generation. It is a bilateral disorder, associated with relatively thick, mid-stromal, lattice-like lines that extend from limbus to limbus (see Fig. 63.24).186,187 Recurrent erosion is relatively infrequent and does not occur in all affected cases. The condition presents between the ages of 60 and 80 years. Occasional unilateral cases have been reported.188 The gene has not yet been mapped.
Other late-onset forms of the BIGH3-related lattice dystrophies have been described, including LCD IIIA, which is an autosomal dominant disorder characterized by recurrent corneal erosions and the onset of visual failure between the fourth to sixth decade. Lattice-like lines are found in the anterior stroma and are characteristically thick and ropy, similar to the lines of LCD III; these ropy lines run from limbus to limbus. This is intriguing because, although it occasionally may be seen in LCD I, usually the peripheral 1 to 2 mm of stroma is clear of dystrophic change in BIGH3-related dystrophies. Amyloid deposits are found in the anterior to mid-stroma, including deposits between the Bowman layer and the stroma-again resembling those found in LCD III.189 Mutations have been demonstrated in BIGH3 at codon Pro501Thr, and the mutated protein colocalized with amyloid deposits in corneal specimens.190,191 However, this mutation has been noted in normal individuals as old as 85 years of age,192 and the factors governing its expression are not understood. Other mutations giving rise to this phenotype are cited in Appendix 1.
Stewart and colleagues193 described a variant of LCD IIIA with onset between the fourth and fifth decades, due to mutations at Asn622His and His626Arg of exon 14 of BIGH3. This has been termed LCD IIIB.3,194 Affected members exhibited marked asymmetry between the two eyes, with lattice changes occurring in those corneas that had suffered recurrent erosions. The authors emphasized that keratoepithelin is expressed during corneal wound healing125 and is inducible by TGF-(, which plays a significant role in epithelial wound healing.195 This observation is important, because it implies that, whereas the mutations may be directly responsible for defective epithelial adhesion, the lattice-like stromal lines may depend on a reactive response to the erosions, possibly involving overexpression of the mutated protein by the epithelium in response to injury. This may have implications for clinical management.
An intermediate LCD I/IIIA phenotype also is described; it presents late, with symptoms arising between the third and fifth decades of life. Patients with the Thr538Arg mutation experience corneal erosions from the teenage years on, and amyloid deposits, mainly subepithelial, have been demonstrated in a keratoplasty button from a 17-year-old patient.135 In two families with a Gly623Asp mutation, patients experienced red, painful eyes at roughly the third to fourth decade, with or without the occurrence of corneal erosions. Very tiny, discrete, subepithelial linear deposits are found in the anterior stroma, leaving the remaining stroma clear. A case of LCD I/111A also was identified from Sardinia, with a delPhe540 mutation, delayed onset, and with erosions occurring from the third decade of life. Again, tiny, branching, subepithelial deposits and an otherwise entirely free stroma were present. The phenotype originally was reported as Reis-Bücklers dystrophy in an earlier report.196 In another family with a His626Pro mutation, corneal changes included a dense stromal haze in addition to lattice lines. Histopathology demonstrated the presence of multiple, fusiform, stromal amyloid deposits. Also, a novel Leu518Arg mutation in an Italian patient was associated with a severe phenotype, with surgery required at the age of 47 years. Subepithelial and stromal amyloid deposits were found.
Deep lattice dystrophy (CDL-deep)135 is an interesting phenotypic variant of the TGFBI dystrophies, described in three families (from Andria, Italy) having a Val631Asp mutation. The mutation is thought to have derived in a common ancestor. The first clinical symptoms appear between 45 and 50 years of age, with photophobia (sometimes followed by erosions) and later visual loss. The earliest corneal features are stellate pre-Descemet opacities associated with round, posterior indentations of the Descemet membrane. This is a distinctive clinical sign, first reported in the polymorphic degeneration phenotype.197,198 Later, lattice lines appear in the mid stroma, suggesting a posterior to anterior progression (the reverse of events seen in LCD I). Asymmetry is common, and the dystrophy may appear to be unilateral in its earliest stages. The anterior stroma barely is affected, and the epithelium is intact. PK has been performed at around 50 years of age.
Polymorphic Corneal Amyloidosis
Eifrig and colleagues described a similar phenotype in a six-generation family, originating from Germany and having a dominant bilateral condition that they termed polymorphic corneal amyloidosis.199 Lesions were gray-white, polygonal, and refractile, resembling chipped ice in appearance and occupying the posterior more so than the anterior stroma (see Fig. 63.24). Occasional, overlapping, linear opacities formed small focal networks. Symptoms of poor night vision with halos and glare, blurring, dryness, or foreign-body sensation and photophobia were reported between the ages of 28 and 40 years. Corneal erosions did not occur. Keratoplasty was performed between the ages of 33 and 59 years, and deposits were shown to contain amyloid. In this TGFBI dystrophy, nine affected patients showed a mutation in BIGH3 (Arg546Asp), whereas three other family members (aged 11 to 15 years) with this mutation were free of corneal signs. A three-generation family having similar clinical features and amyloid or hyaline stromal deposits was reported by Pillat.200
Polymorphic Amyloid Degeneration
Thomsitt and Bron described a bilateral, polymorphic condition of the corneal stroma characterized by small dots, flecks, and linear and stellate lesions sometimes resembling snowflakes, ranging 12 to 106 (m in diameter. These lesions occasionally were accompanied by branching, lattice-like lines similar to those seen in LCD. In six out of the nine patients observed, lesions were confined to the posterior stroma or decreased in number anteriorly. They occupied a discoid or rarely annular zone of the stroma (Fig. 63.25). The most posterior lesions indented the Descemet membrane in a characteristic way to produce a dark spot on specular reflection with an underlying normal endothelial mosaic. In one case, the distribution was pan-stromal and, in another, it occupied only the anterior stroma. Corneal sensation and thickness was normal, and the perilimbal stroma was always clear. Vision was rarely affected. This condition occurs in both sexes, and presents between the ages of 51 and 76 years. It was originally described as a dystrophy,197 but was renamed as polymorphic amyloid degeneration by Mannis and colleagues198 who identified the amyloid nature of the lesions and stressed the lack of familial history. The condition can be regarded as a phenocopy of polymorphic amyloid dystrophy, but with a later presentation and few if any symptoms. A similar condition was described by Strachan.201
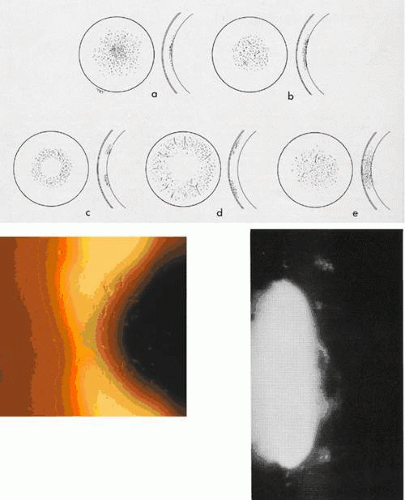 Figure 63.25. Polymorphic amyloid degeneration. A: Schematic of the distribution of polymorphic opacities in this disorder, showing ring-shaped and discoid arrangements. B: A slit-lamp view of the snowflake opacities illustrate their resemblance to those of polymorphic amyloid dystrophy. C: A characteristic feature is the manner in which posterior opacities indent the Descemet membrane without influencing the integrity of the underlying endothelium. (Figures A and C reproduced with permission from Thomsitt J, Bron A: Polymorphic stromal dystrophy. Br J Ophthalmol 1975;59:125–132.) |
A further, late-onset form of deep lattice dystrophy (LCD IV), caused by a Leu527Arg mutation, was described by Fujiki and colleagues.113,202 A sporadic case of LCD occurring in a 68-year-old man was designated as an atypical form of LCD IV on the basis of late onset, lack of corneal erosions, and the presence of amyloid deposits in the mid and posterior stroma (203). A mutation was found at Asn544Ser.
Other variants of LCD also have been described. A variant associated with an Ala546Thr mutation in TGFBI has been called LCD I-like, or French LCD IIIA by Dighiero and colleagues.173 An LCD I/IIIA intermediate form was associated with mutations in exon 14 of TGFBI (a nine-base pair insertion at nucleotide 1885-1886 and a missense mutation at nucleotide 1887).204 An asymmetric variant of LCD with a His626Arg mutation was reported by Dighiero and colleagues and others.135,173 Klintworth has referred to these phenotypes as types V, VI, and VII, respectively.3
Forms of Lattice Corneal Dystrophy Unassociated with BIGH3 Mutations
Type II Lattice Dystrophy (Meretoja Syndrome)
LCD II is an autosomal dominant form of LCD associated with the Meretoja syndrome, the Finnish type of familial systemic amyloidosis (familial amyloid polyneuropathy, FAP type IV).205,206,207,208 The lattice-like corneal changes are of later onset and more peripherally located than in LCD I, and patients do not suffer erosive attacks.
It is caused by mutations in the gelsolin gene (Asp187Asn and Asp187Tyr), which is located on chromosome 9q34 (see Table 63.1).209,210,211,212,213,214,215 Paunio and colleagues have demonstrated that cells of neural origin have a greater ability than non-neural tissues to process mutant gelsolin into an amyloid precursor protein.216 This may be relevant to the clinical manifestations of the disorder.
Patients exhibit a progressive cranial and peripheral neuropathy, with dry skin, blepharochalasis, protruding lips, lagophthalmos, and a mask-like facies. Lattice-like lines, thicker, more peripheral, and less numerous than those of LCD I, appear in the corneal stroma after the age of 20 years, spreading centripetally from the limbus. The central cornea is relatively spared, so that vision usually is not affected before the age of 65 years. However, corneal changes may be exacerbated by lagophthalmos. A reduction occurs in the number of long, sub-basal nerve plexus fibres and an associated reduction in corneal sensitivity, which may explain the occurrence of dry eye in some patients.217 Although corneal grafting is rarely indicated, keratoplasty has been associated with a neurotrophic, persistent epithelial defect, reflecting a reduced corneal sensitivity. Erosive symptoms may be a presenting feature in the third decade of life.
Tsunoda described a nonfamilial form of amyloid A amyloidosis in conjunction with lattice dystrophy, polyneuropathy, and a profound autonomic neuropathy.218 The disorder showed some similarities to the Meretoja syndrome but appears to be distinct.
The amyloid deposit in the Meretoja syndrome is composed of a 71-amino acid, mutant gelsolin protein. Amyloid is deposited in the nerves, arteries, skin, heart, and kidney; in the eye, it is found in the blood vessels of the sclera, choroid, and lacrimal gland and in the perineurium of the ciliary nerves.207 In the cornea, it accumulates in the stroma and in the subepithelium where it is immunolocalized by anti-gelsolin antibody.107,219
Lattice Dystrophy with Myelopathy
Another systemic association of LCD was observed in our laboratory (AJB and SA), in which lattice dystrophy cosegregated as a dominant trait with a myelopathy in a father and two daughters. Asymptomatic LCD was detected in the daughters in the third decade (Fig. 63.26). The father experienced visual symptoms from the age of 38 years, due to chronic glaucoma and corneal changes. He underwent combined corneal grafting and cataract extraction at the age of 51 years. The neurologic presentation in this family was initially that of a distal spinal muscular atrophy, with weakness and wasting of the hands and upper limbs and some sensory involvement. Magnetic resonance imaging (MRI) demonstrated antero-posterior narrowing of the cervical canal with local cord atrophy in the absence of compression. The most severely affected daughter developed a more diffuse spinal cord syndrome, with lower limb spasticity and dorsal column involvement. Neurologic changes were more advanced in the daughters than in the father. Corneal signs consisted of stromal flakes and delicate lattice-like lines in the affected sisters, with more advanced changes in the father. The father’s stroma showed posterior, flake-like lesions that indented the Descemet membrane, much as they do in polymorphic amyloid degeneration.197,220 Stromal amyloid deposits were demonstrated in graft material from the affected father. Keratoepithelin was found in increased amounts in the stroma, although no mutation was detected in the reading frame of the BIGH3 gene or in the SOD-1 (superoxide dismutase) and SMN (survival motor neurone) genes that have been implicated in certain motor neurone disorders.
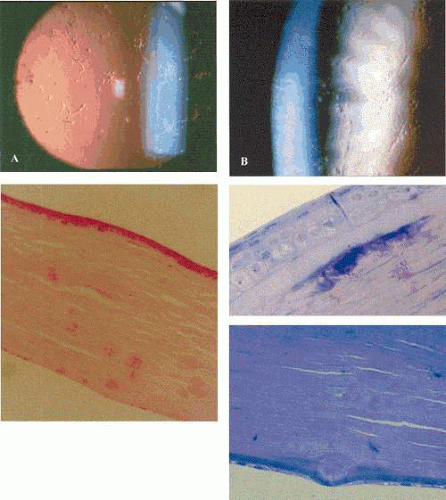 Figure 63.26. Lattice dystrophy with myelopathy. A: Slit-lamp picture shows fine lattice-like lines against the red reflex in an affected patient. B: Detail of lattice-like lines, seen in retroillumination. C: Stained LM section shows that a greater proportion of deposits (stained pink) are found in the posterior stroma. D: Semi-thin, toluidine blue-stained section shows a deposit subjacent to an intact Bowman layer. E: Semi-thin section shows a deposit that has pushed the Descemet membrane backward, thinning the overlying endothelium. This is a typical feature of this disorder and of polymorphic amyloid degeneration (from Talbot Akhtar Doneghy and Bron, in preparation).[JW46] |
Macular Dystrophy
Macular corneal dystrophy (MCD) is an autosomal recessive dystrophy caused by a defect in carbohydrate sulfation. It gives rise to diffuse corneal clouding from childhood or adolescence and to progressive visual failure. Different immunophenotypes exist.
MCD maps to chromosome 16q23.1-23.2221,222 and is caused by mutations in a carbohydrate sulfotransferase gene (CHST6) that lead to undersulfation of the glycosaminoglycan (GAG) keratan sulfate (KS) (see Table 63.1 and Appendix 1).223,224,225 Expression of mutant genes in cornea and cartilage leads to a fall in KS content in both these tissues.
Klintworth and colleagues discovered that immunodetectable KS was deficient in the serum and corneas of some but not all patients with MCD,226,227,228, giving rise to three immunophenotypes, MCD I, Ia, and II.229,230 In MCD I, the most common variety, almost no detectable sulphated keratan sulphate is present in serum or cornea. In MCD Ia, sulphated KS is absent from the serum but is detectable in the cornea within keratocyte accumulations.231 In MCD II, normal amounts of sulphated KS are present in both the serum and corneal intracytoplasmic and stromal deposits. The three types are biomicroscopically identical.
These studies indicated the involvement of cartilage in MCD I, in which the content of immunoidentifiable, sulphated KS in cartilage was about 800 times less than normal.232 However, histopathologic lesions have not been identified in cartilage or other nonocular tissues,3 and it appears that cartilage is involved at a metabolic, but not a clinical level.227,228
Early linkage studies indicated that MCD I was probably at the same locus as MCD II,221 supported by the demonstration of different immunophenotypes in the same families231,233 and even in the same sibships.222 These observations have since been borne out by mutational studies that show types I and II to be allelic disorders.
In MCD type I, mutations affecting the coding region of CHST6 inactivate the enzyme and result in the synthesis of undersulfated KS proteoglycans in the cornea. The CHST6 gene encodes an enzyme that is a member of the GST sulfotransferase family. Three of the GST family (CHST4, CHST5, and CHST6) occupy a cluster on the long arm of chromosome 16. Although CHST6 is expressed in the cornea and brain, no cerebral manifestations accompany mutations of this gene, probably because of the compensatory activity of other sulfotransferases.
The CHST6 enzyme catalyses sulfation the 6-position of N-acetyl glucosamine and possesses a 3′phosphosulfate (PSB) and a 5′phosphate binding (PB) site for a high-energy sulphate donor (PAPS), bordering a pocket for the N-acetyl glucosamine of KS. These sites bring the sulfate donor into proximity with the KS sugars to facilitate sulfation.234 Most of the CHST6 mutations giving rise to MCD I are homozygous mutations located at or near the binding sites or the binding pocket of the gene; that is, the Pro204Gln and Arg205Leu mutations are in the 3′PSB domain and the Arg211Trp mutation is adjacent to it. The Arg177His mutation is in the sequence between the 5′-PB domain and the 3′ PB domain. Other mutations do occur outside the binding regions; for example, Glu274Lys,223 Pro31Ser,185 Ala217Thr,223 and the Ala217Thr mutation still leads to a failure of synthesis of sulphated KS.223[JW6] Compound heterozygous mutations also are described.234
MCD II is caused by mutations upstream to the coding region of the gene and lead to a loss of corneal epithelium-specific CHST6 expression,223 and to a lack of sulfotransferase activity in the epithelium but not the corneal stroma or serum. It is believed that undersulfated KS of epithelial origin contributes to the stromal deposits.
Atypical forms of MCD also have been described.234 One patient with an Arg211Trp mutation showed absent serum KS but interlamellar accumulations of KS in the corneal stroma; this case was regarded as an atypical form of MCD type I. Two other patients who had originally been diagnosed as type II, despite mutations in the coding region, showed absent KS staining in the stromal matrix but positive staining in the deposits. One at least had normal serum KS levels.
The biochemical basis of MCD. Proteoglycans (PGs) are important macromolecular components of the extracellular matrix, including the cornea. They are composed of a protein core attached to multiple GAG side chains. KS is the most abundant GAG of the cornea, making up about 50% of corneal GAGs. It is found in the PGs lumican, keratocan, and mimican. In KS, sulfation occurs in particular, at terminal lactosaminoglycan moieties. The high negative charge at the surface of the molecule contributes to the gel properties of proteoglycans, such as the swelling pressure of the corneal stroma. Cartilage is also rich in KS, and cartilage metabolism is the major contributor to serum levels of KS in normal subjects.
The search for the basis of MCD is an exciting story of medical detection, and is reviewed in detail by Klintworth, who played a major role in the process of discovery.3 Early histochemical studies229,231,235 suggested that the corneal deposits in macular dystrophy contained GAGs and suggested a defect in GAG degradation, as occurs in the systemic mucopolysaccharidoses. A role for keratocytes was implied by their known ability to synthesize GAGs.236,237 KS appeared to be the most likely candidate for the accumulated product found in these deposits, based on the selective resistance of deposits to enzymatic digestion and their positive staining with PAS and with Alcian blue in controlled conditions at low pH.229
An important observation was the low level of KS synthesis by organ-cultured MCD corneas (229) and of lumican, the major KS-containing proteoglycan. This implied a defect in an enzyme required for lactosaminoglycan sulfation.229,238 Corneas with MCD synthesized altered proteoglycans and lactosaminoglycan-containing glycoproteins,239 but produced an excess of variably sulphated glycopeptides.240,241 Both cartilage and cornea contained reduced KS side chains in MCD.242
Organ-culture studies of MCD corneas suggested a fundamental defect in KS sulfation in at least one form of MCD (type I), and hence in the biosynthesis of KS-containing PGs.228,229,239 This was thought to lead to defective KS PG synthesis in MCD corneas,238,243 probably due to a defect in a specific sulfotransferase that attaches sulfate moieties to the lactosaminoglycan of nonsulfated KS.227,228,238 Although the serum levels of sulfotransferases were normal in MCD,244,245 the level of the sulfotransferase, N-acetylglucosamine 6-O-sulfotransferase (GlyNAc6ST) in corneal extracts was reduced, which identified the biochemical basis of MCD.246
MCD causes a diffuse stromal corneal clouding that usually presents in adolescence but may appear in infancy or late adult life.247,248 Changes in infancy are not sufficient to induce either amblyopia or nystagmus (Fig. 63.27). Unlike the situation in the BIGH3 dystrophies, the corneal changes extend to the corneal limbus, without a peripheral clear zone. With time, the stromal clouding increases in density and is associated with the appearance of dense, mainly superficial “macules” that elevate the corneal surface and contribute to the visual loss. These macules give rise to the disorder’s name. They appear to spare the most peripheral region of the cornea, suggesting that these larger opacities may be subject to the same constraints that leave the peripheral stroma clear in the TGFBI dystrophies. Thinning of the cornea occurs, which is attributed to a decreased separation of collagen fibrils249 and possibly reflects the altered swelling properties of the stromal GAGs.
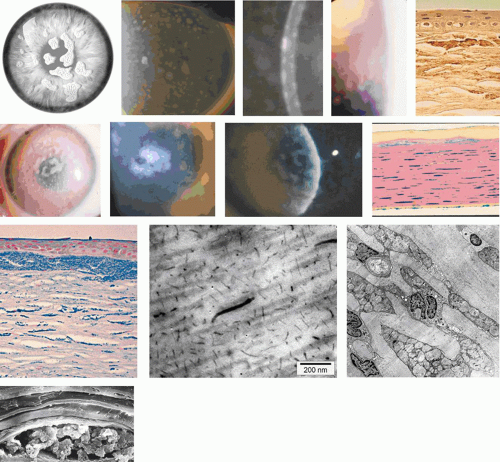 Figure 63.27. Macular corneal dystrophy (MCD). A: Schematic drawing shows the distribution of opacities in MCD. Note that the stromal haze extends to the periphery of the cornea. B-F: Slit-lamp features of MCD. B: Survey picture of MCD in an adult, showing multiple small macular opacities situated within a general stromal haze. The macules extend to the corneal periphery. In slit section (inset), it can be seen that although the macules are most defined anteriorly at the level of the Bowman membrane, similar patches of opacity are present within the endothelium (Reproduced with permission from Aldave AJ, Yellore VS, Thonar EJ, et al[JW47]. Novel mutations in the carbohydrate sulfotransferase gene (CHST6) in American patients with macular corneal dystrophy. Amer. J Ophthalmol 2004;137:465). C: In another patient, the macules are more developed, but do not extend to the far periphery. D: A high-power view of the perilimbal cornea in this patient shows how the ground-glass stromal clouding extends beyond the most peripheral macules. E: High-power view of a large macule, elevated above the corneal surface. F: A broad, slit-section of the cornea in a further patient demonstrates the surface irregularity caused by superficial macules. G: Light micrograph of an MCD cornea showing the accumulation of mucopolysaccharide within swollen keratocytes and stromal deposits. The Bowman membrane is partially destroyed by the deposits closest to the surface, which probably correspond clinically to MCD macules. Note the involvement of the endothelium in this micrograph (Alcian blue stain). H: Light micrograph shows a more extensive subepithelial deposit, which has destroyed the Bowman membrane. It is overlain by a fine pannus (Hale’s colloidal iron; courtesy B McDonald). I: Light micrograph shows anterior stromal deposits staining positively with an antibody directed against keratan sulfate, in MCD type II. (courtesy GN Klintworth). J: TEM of stroma from a patient with MCD, stained with cuprolinic blue, shows the presence of abnormally large proteoglycans filaments distributed among the collagen fibres. K: TEM showing enlarged stromal fibroblasts packed with abnormal proteoglycans. L: SEM of proteoglycan deposits in the corneal stroma. (Figures K, L, courtesy of GN Klintworth).
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|