Inborn Errors of Metabolism
Donald P. Sauberan MD
Scott E. Olitsky MD
Leonard Nelson MD
An inborn error of metabolism is a clinical disorder caused by a genetic inability to produce the full complement of a given functioning protein in its normal configuration. Many of these disorders are rare events with often devastating ocular manifestations. Because of the high incidence of these ocular manifestations, the ophthalmologist is often involved in the care of these patients. Also, there is an increasing diagnostic role for the ophthalmologist in detecting subtle, early, and occasionally pathognomonic lesions to aid in the diagnosis of these rare disorders.
Disorders of Amino Acid Metabolism
Homocystinuria
Homocystinuria is an inborn error of metabolism of the sulfur-containing amino acids. Carson and Neill first detected homocystinuria using urine chromatography while systematically searching for metabolic abnormalities in mentally retarded institutionalized persons in Northern Ireland.1 Simultaneously and independently, Gerritsen and associates in Madison, Wisconsin, found the same metabolic defect in an infant with mental retardation and failure to thrive.2
Homocystinuria is an autosomal recessive disorder caused by a deficiency in the pathway that converts the sulfur atom in methionine to the sulfur atom in cysteine.3 The most common enzymatic defect is a mutation of cystathione β-synthase, which normally converts homocysteine into cystathione. The resulting defect in this biochemical pathway causes accumulation of homocystine and methionine with increased concentrations of these amino acids in blood and urine.4 At least seven other causes of homocystinuria have been described (OMIM). The cystathione β-synthase genetic mutation has been mapped to chromosome 21 (21q22.3).
There are four organ systems that show major involvement in patients with homocystinuria: skeletal, central nervous, vascular, and the eye (Table 54A.1). Excessive height and long, thin extremities are common skeletal findings in the patient with homocystinuria. These findings are similar to those of the Marfan syndrome. The most consistent skeletal abnormality is osteoporosis, which may lead to vertebral collapse.5 Developmental delay occurs in approximately 65% to 80% of cases and may be progressive. Seizures and psychiatric disturbances can also occur. Thrombotic vascular occlusions constitute the main threat to survival in patients with homocystinuria. Large and small arteries and veins may be affected. Therefore, premature deaths from myocardial infarctions, pulmonary emboli, and cerebrovascular accidents are not uncommon. Approximately 50% of untreated patients die by age 25.
Anesthesia may present significant risks to the homocystinuric patient.6,7,8 Mudd and colleagues documented 25 postoperative thromboembolic events in 586 homocystinuric patients undergoing surgical procedures, six of them fatal.9 Medications that may predispose these patients to a hypercoagulable state, such as oral contraceptives, should also be avoided.10
The ophthalmologist can often help diagnose homocystinuria. Ectopia lentis is the ocular hallmark of homocystinuria and can be detected in approximately 90% of patients.11,12 Ectopia lentis is an acquired and progressive abnormality in this disorder. The dislocation is bilateral, with the lens usually migrating either inferiorly or inferonasally. The causes of ectopia lentis are summarized in Table 54A.2.
TABLE 54A.2. Differential Diagnosis of Ectopia Lentis | ||
|---|---|---|
|
The lens in homocystinuria is much more mobile than in the Marfan syndrome. This may be related to the clinical observation of progressive irregularity of the zonular fibers and the appearance of a fringe of white zonular remnants at the equator of the lens and on the surface of the ciliary body (Figure 54A.1).13,14 Henkind and Ashton first reported histopathologically the ocular findings in four eyes of three homocystinuric patients. They found the zonular fibers to be deficient adjacent to the lens. These zonules had recoiled to the surface of the ciliary body and were matted and retracted into a feltwork that fused with a greatly thickened basement membrane of the nonpigmented epithelium. The greatly thickened basement membrane overlying the ciliary body in homocystinuria has subsequently been shown by electron microscopy to be composed of degenerate zonular material.15 In addition, Ramsey et al noted that the degree of zonular abnormality was related to age: the younger the patient, the more normal-appearing zonular fragments composed of oriented filaments that could be identified.15 The zonular fibers are composed of glycoproteins with a high concentration of cysteine, which may explain their susceptibility to abnormal function in homocystinuria.16,17
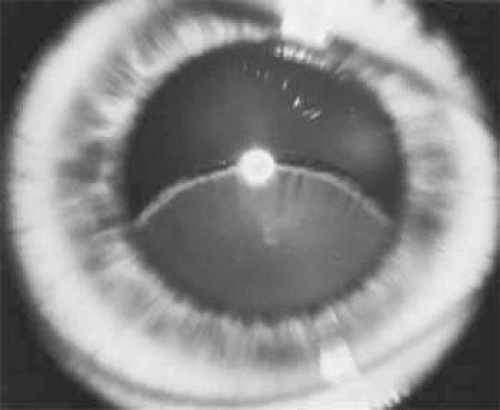 Figure 54A.1. Inferior dislocation of the lens in a homocystinuric patient. Note the absence of most zonular fibers. (Nelson LB, Maumenee IH: Ectopia lentis. Surv Ophthalmol 27:143, 1982) |
Myopia is another common ocular finding and may precede the detection of ectopia lentis by several years.17 The development of progressive lenticular myopia is often the first sign of a lens dislocation. Retinal detachment is usually a complication of lens surgery, although it may occur spontaneously.12
Other ocular findings associated with homocystinuria are listed in Table 54A.3.
TABLE 54A.3. Ocular Findings Associated with Homocystinuria and the Frequency of Their Occurrence | ||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||
One treatment of homocystinuria to correct the metabolic defect has involved a low-methionine and high-cystine diet, which has not been uniformly successful.19,20,21,22,23 This diet has not only failed to normalize completely the abnormal biochemical findings, but it is difficult to prescribe (all eggs, meat, and cow’s milk are forbidden). When this diet is prescribed to infants, failure to gain weight may be a serious complication.24
Another approach to therapy is supplementation with coenzymes. Pyridoxine (vitamin B6) is the coenzyme necessary to activate cystathionine beta-synthetase.25 Barber and Spaeth were the first to demonstrate that certain homocystinuric patients respond biochemically to oral pyridoxine with lowered plasma concentrations of homocystine and methionine.26 A different mutation in the cystathionine beta-synthase gene is responsible for the observed difference in the individual response to pyridoxine.27,28 Homocystinurics who are B6-responsive have a less devastating clinical course, with a higher average IQ and less frequent major clinical abnormalities. In addition, B6-responsive individuals may show a decrease in the incidence of clinical abnormalities when treated with pyridoxine.9,29 Approximately 50% of patients with homocystinuria are responsive to B6.
Early identification of patients with homocystinuria is essential so that early and effective treatment can be provided. Spaeth and Barber first described a sodium-nitroprusside test to screen for homocystine in the urine.30 Because the test results are positive in many conditions in which sulfur-containing metabolites are excreted (e.g., cystinuria), urine chromatography or high-voltage electrophoresis is necessary for a definitive diagnosis. Some B6-responsive cases can be missed with current screening protocols.9,31 Thirty-eight states currently have mandatory newborn screening for homocystinuria. However, increased concentrations of methionine may be minimal during the first 3 days of life until there is adequate protein intake (milk feedings). This is especially true in patients who are responsive to vitamin B6, who usually have some residual enzyme activity.4
Albinism
Albinism comprises a heterogeneous group of clinical syndromes exhibiting hypomelanosis based on heritable metabolic defects of the melanin pigment system. All types of albinism are characterized by foveal hypoplasia, nystagmus, photophobia, and decreased visual acuity in addition to absent or decreased melanotic pigment in skin, hair, and eyes (oculocutaneous albinism [OCA]) or in the eye alone (ocular albinism). The term albinoidism refers to hypomelanotic disorders in which the patients do not have nystagmus, decreased visual acuity, or foveal hypoplasia. Eleven disorders have been identified with clinical features of oculocutaneous albinism and four with features of ocular albinism (Table 54A.4). The ocular findings in oculocutaneous albinism are listed in Table 54A.5 (Figs. 54A.2 and 54A.3).
TABLE 54A.4. Classification of Inherited Hypopigmented Conditions With Ocular Involvement | ||
|---|---|---|
|
TABLE 54A.5. Ocular Signs in Oculocutaneous Albinism | |
|---|---|
|
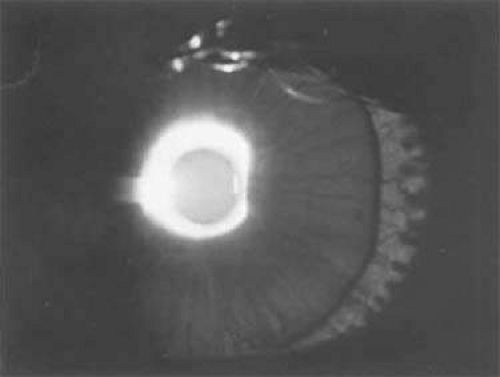 Figure 54A.2. Diffuse transillumination in a diaphanous hypopigmented iris of a child with oculocutaneous albinism. Note the lens edge. |
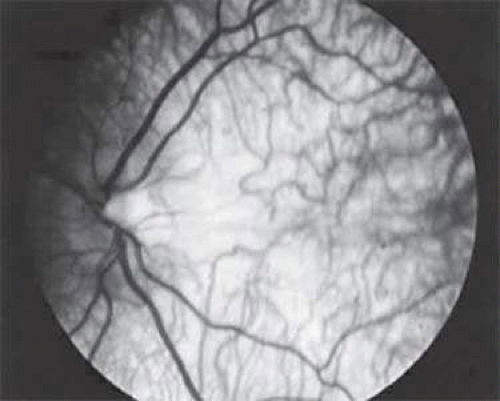 Figure 54A.3. Pigmentary dilution of the retina in oculocutaneous albinism. Note the absence of the macular reflex. Large and tortuous choroidal vessels are easily seen; these vessels are not visible when the retinal pigment epithelium is normal. |
Individuals who are homozygous for genetic mutations responsible for OCA have an abnormal proportion of fibers from the ganglion cells of the temporal retina that decussate to the contralateral cerebral hemisphere; this can be demonstrated by monocular visual evoked potential asymmetry.32,33 This may play a role in the high incidence of subnormal binocular vision and strabismus in albinos. This abnormality can be detected early in life and may be helpful in making the diagnosis of OCA during the neonatal period.34
Four forms of OCA have been identified, each mapped to a different genetic locus. OCA type I represents tyrosinase-negative albinism and has been mapped to chromosome 11 (11q14-q21). There is a deficiency in the catalytic activity of tyrosinase, which is involved in at least three steps in the melanin biosynthetic pathway.35,36 In OCA type IA, the affected homozygote is “dead white” at birth, with no discernible pigment, and has complete absence of tyrosinase activity. In the yellow mutant form of OCA (type IB), there is reduced activity of tyrosinase. Some pigmentation of the skin forms and the hair develops a yellow coloration.
Tyrosinase-positive OCA (type II) has been mapped to chromosome 15 (15q11.2-q12) and is the most prevalent type of albinism in the world.37 The mutated protein (OCA2) is a transmembrane polypeptide that may play a role in transporting small molecules such as tyrosine, a precursor of melanin.38,39 Affected individuals show some degree of pigmentation and have less profound clinical manifestations than those with OCA type I. Brown oculocutaneous albinism is thought to be an allelic variant of OCA2.
In OCA type III, a mutation occurs in the tyrosinase-related protein-1 (TYRP1) locus located on chromosome 9 (9p23). While its specific function is unknown, this mutation of TYRP1 results in dysregulation of tyrosinase activity. This promotes the synthesis of brown melanin instead of black melanin.40 Affected individuals have reddish hair and red-brown skin tone and are capable of developing a faint tan with sun exposure.41
OCA type IV is caused by a mutation in the membrane-associated transporter protein (MATP) gene, located at 5p13.3. Patients with OCA type IV are phenotypically similar to those with OCA type II.
There are two systemic disorders that need to be considered in patients with a diagnosis of oculocutaneous albinism. All patients with OCA should be questioned about a history of bleeding because of the association of albinism with a hemorrhagic diathesis, the Hermansky-Pudlak syndrome.42 Four subtypes of Hermansky-Pudlak syndrome have been identified, depending on the genetic mutation involved. All consist of oculocutaneous albinism along with lysosomal accumulation of ceroid lipofuscin and defective platelets. The hemostatic defect in the Hermansky-Pudlak syndrome is typically mild. The most common manifestations are gingival bleeding, epistaxis, and easy bruisability.43 More serious bleeding episodes have followed surgical procedures, especially when aspirin was used to control postoperative discomfort.44 The Hermansky-Pudlak syndrome is the most common form of albinism seen in Puerto Rico. A deficiency of platelet dense bodies, as observed by electron microscopy, is the most reliable method of diagnosing this syndrome.45
Chediak-Higashi consists of oculocutaneous albinism along with immune system defects leaving patients susceptible to recurrent infections. These immune system defects include natural killer cell dysfunction, impaired neutrophil chemotaxis, and the defective gene is the lysosomal trafficking regulator gene (LYST) at chromosome 1q42-43. Death often occurs within the first decade, either due to overwhelming infection, or due to an accelerated lymphoma-like phase. Bone marrow transplantation may be used to alleviate the immunologic disorders.
Nettleship-Falls albinism (X-linked ocular albinism) is caused by a defect in the G protein-coupled receptor 143 (GPR143).46 This defect has been localized to the Xp22.3 region. The ocular findings in ocular albinism are similar to those in OCA. However, in patients with ocular albinism, the pigmentary dilution of the eye may be subtle. In whites, there is usually iris transillumination. In blacks, the iris often does not transilluminate and the retina is usually moderately pigmented.47 Regardless of the pigmentation of affected patients, all have foveal hypoplasia. In affected persons with ocular albinism, the cutaneous pigmentary dilution is much less noticeable than is the ocular involvement. The skin pigmentation in these patients typically falls within the normal range. However, when these affected persons are compared with their nonaffected siblings, they typically have a lighter complexion.48 Histopathologic examination of the clinically normal skin in affected males and carrier females has revealed the presence of macromelanosomes.49 Female carriers often show a mud-spattered appearance of the fundus with hyperpigmented streaks as well as iris transillumination defects. These clinical signs, in addition to skin biopsy, can be used to help in genetic counseling for families at risk.50,51
Diagnosis of albinism is generally made based on clinical examination. If needed, DNA testing is available for some forms of albinism and electron microscopic evaluation of the skin of patients with suspected ocular albinism may be helpful. In addition, a visual evoked potential may be useful to detect the misrouting of ganglion cells of the temporal retina typically seen in these disorders. This abnormality can be detected early in life and may be helpful in making the diagnosis of OCA during the neonatal period.
Treatment includes the correction of refractive error when necessary and the use of low vision aids if needed. Patients should be referred to a dermatologist for proper skin care. Genetic counseling is helpful due to the hereditary nature of these disorders. Patients in whom Hermansky-Pudlak or Chediak-Higashi syndromes are possible should also be referred for appropriate consultation.
Tyrosinemia
There are five clinical syndromes in which elevated serum and urinary tyrosine levels and their metabolites can be detected: (i) tyrosinemia secondary to systemic disease (liver damage, pernicious anemia, vitamin C deficiency); (ii) neonatal tyrosinemia, a transient benign process resulting from immature parahydroxyphenylpyruvate oxidase producing high serum tyrosine levels in premature infants; (iii) tyrosinemia associated with hepatorenal disease (hereditary tyrosinemia, tyrosinemia type I, which is a rapidly fatal disease of infancy), methionemia, aminoaciduria, and glycosuria; (iv) tyrosinemia associated with ocular but not hepatorenal disease (essential tyrosinemia, tyrosinemia type II); and (v) tyrosinemia without hepatorenal or ocular disease.
Type I tyrosinemia is caused by a defect in fumarylacetoacetate hydrolase (chromosome 15q23-q25), the last enzyme in the tyrosine catabolism pathway.52 An accumulation of succinylacetone occurs that reacts with other amino acids and proteins. Acutely, this produces jaundice and elevates liver transaminases. Patients who survive beyond infancy are at high risk for the development of hepatocellular carcinoma. This risk has been prevented in some children who have undergone neonatal liver transplantation.53 Pharmacologic treatment with 2-(2-nitro-4-trifluoromethylbenzyl)-1,3-cyclohexanedione (NTBC) has changed the outcome of this disorder dramatically. 90% of patients will respond to NTBC. Death usually occurs in the first decade of life.
Tyrosinemia type II (Richner-Hanhart syndrome) is a rare congenital error of metabolism characterized by a triad of dendriform keratitis, hyperkeratotic lesions of the palms and soles, and mental retardation (Figures 54A.4 and 54A.5).54,55,56,57,58,59,60 The enzymatic basis of the syndrome is a defect in soluble hepatic cytosol tyrosine aminotransferase, whose gene has been mapped to chromosome 16 (16q22.1-q22.3).56,61,62
 Figure 54A.4. Subepithelial corneal opacification in tyrosinemia type II. (Courtesy Marian Macsai, MD) |
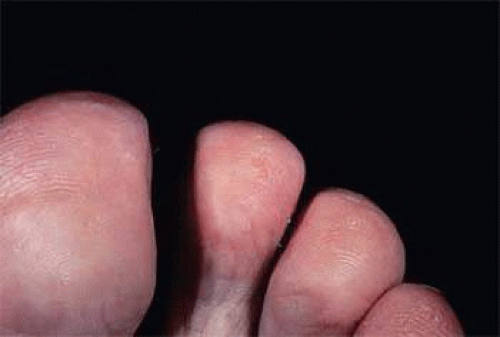 Figure 54A.5. Hyperkeratotic lesion of the soles in a patient with tyrosinemia. (Courtesy Marian Macsai, MD) |
Many of the early reported cases of this disease were in patients who were the product of consanguineous marriages, suggesting a possible autosomal recessive inheritance.56,59,60,63,64
The diagnosis of tyrosinemia type II may be confirmed by amino acid analysis of the blood and urine, which shows an increase of tyrosine and its metabolites only. Ocular symptoms of photophobia and lacrimation in both eyes usually appear during the first few months of life. The affected patient has bilateral subepithelial central corneal ulcers with herpetiform dendritic branching, which may develop with time into round whitish opacities with superficial new vessel formation.60 The corneal ulceration probably results from accumulation of intracellular crystals, presumably tyrosine. These crystals have been shown to enlarge, lacerate, and rupture the cell; attract lysosomal enzymes; and trigger the inflammatory cycles.55,65
A low-tyrosine, low-phenylalanine diet has been used successfully in several patients with tyrosinemia type II, with rapid resolution of the clinical symptoms and signs.56,59,66,67,68,69 The keratitis found in tyrosinemia type II can be distinguished from herpes simplex keratitis by its morphologic appearance, bilateral presentation, lack of response to antiviral therapy, and associated systemic findings.54 Therefore, an infant or young child with bilateral dendritic keratitis should undergo serum and urinary evaluation for elevated levels of tyrosine and its metabolic byproducts. Early dietary treatment can reduce the development of mental retardation.
Tyrosinemia type III is due to a defect in the enzyme 4-hydroxyphenylpyruvate dioxygenase, which has been mapped to chromosome 12 (12q24). Liver biopsy in affected individuals has been histologically normal, and mental retardation may be present.70
Cystinosis
Cystinosis is an inherited, autosomal recessive disorder of amino acid metabolism characterized by the deposition of cystine crystals in various tissues such as the eye, bone marrow, lymph nodes, and internal organs.71 Cystine is the disulfide of the amino acid cysteine. Cystinosis is a lysosomal storage disorder that results from impaired transport of cystine from cellular lysosomes. It differs from other lysosomal diseases because the main enzymatic function of lysosomes, acid hydrolysis, does not play a role in cystine deposition. Plasma levels of cystine are below saturation level, whereas intracellular concentrations are elevated in polymorphonuclear leukocytes, macrophages, and fibroblasts, further indicating the cellular nature of the defect responsible for this disease.
All forms of cystinosis are caused by a defect in the CTNS gene, which is located at chromosome 17p13. This gene encodes cystinosin, a 367 amino acid integral membrane protein that transports cystine out of the lysozome.72 The resulting decrease in cystinosin leads to an elevated level of intracellular cystine, which leads to tissue deposition. The three clinical phenotypes of cystinosis are formed by different mutations in the CTNS gene, which vary based on the amount of residual cystinosin activity.73
All three clinical subtypes of cystinosis have ocular involvement (Table 54A.6).74 The infantile, or nephropathic, variety is the most severe form of cystinosis.75 The gene has been mapped to the short arm of chromosome 17 (17p13).76 During early childhood, affected patients develop polyuria and polydipsia as a result of impaired renal tubular water reabsorption. This is followed by growth retardation, renal rickets, metabolic acidosis, progressive renal failure, and, ultimately, death from uremia before puberty.
TABLE 54A.6. Some Distinguishing Characteristics of the Three Major Types of Cystinosis | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Children with nephropathic cystinosis usually develop severe photophobia within the first few years of life. Slit-lamp examination discloses diffuse scintillating, tinsel-like crystals of the cornea, conjunctiva, and iris (Fig. 54A.6).77,78,79 Initially, the iridescent crystals occupy the full stromal thickness only in the periphery, while centrally the anterior half to two thirds of the stroma is involved. By 16 months of age, corneal crystals are clinically present.80 By approximately 20 years of age, the entire corneal stroma has crystals. In patients with longstanding disease, thinning and focal breaks in Bowman’s membrane may be present and contribute to the development of severe photophobia.81 Corneal sensitivity may be reduced.82
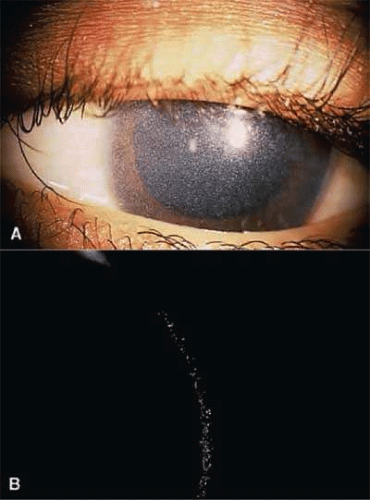 Figure 54A.6. Slit-lamp view of crystal deposition in cystinosis. (Courtesy James J. Reidy, MD) |
The essential fundus abnormality is a patchy depigmentation of the periphery and a finer “salt-and-pepper” disturbance at the posterior pole.75,83 Crystal deposition may also be seen (Fig. 54A.7). Despite the extensive tissue infiltration of cystine crystals, there is no significant visual disturbance.
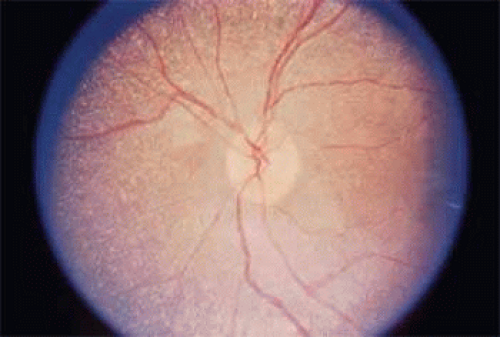 Figure 54A.7. Crystal deposition of the fundus in nephropathic cystinosis. (Courtesy James J. Reidy, MD) |
Cysteamine (beta-mercaptoethylamine) is a cystine-depleting agent used in the treatment of infantile nephropathic cystinosis. It reacts with cystine to form cysteine and a cystine-cysteamine complex, both of which freely exit the lysosome. When given systemically, it has proven successful in retarding glomerular deterioration and enhancing growth, but it does not prevent the development of corneal crystal formation.71,76,84,85,86 Topical cysteamine drops have been shown not only to provide primary prevention of corneal crystal deposition but may also reverse the corneal complications in older patients.89,90
Adolescent cystinosis is a milder form of the disease, appearing in the first or second decade of life.89,90 This type of cystinosis may or may not include rickets, renal failure, photophobia, or retinopathy. All patients have shown the crystalline material in the cornea, conjunctiva, and reticuloendothelial system.
A benign adult form of cystinosis was first described by Cogan and coworkers in 1957.91 The primary clinical distinction between the benign and the nephropathic variants is failure of the former patients to show either retinopathy or renal dysfunction (see Table 54A.6).92 Although affected patients may have mild photophobia, they are frequently diagnosed by routine ophthalmologic examination when the typical corneal crystals are noted.93,94
Idiopathic intracranial hypertension has also been shown to occur in patients with cystinosis at a higher rate than expected.95 Postulated mechanisms include blockage of CSF absorption by the arachnoid villi secondary to cystine infiltration, and hypercoagulability secondary to renal disease.
Gyrate Atrophy
Gyrate atrophy of the choroid and retina is a rare, autosomal recessive, progressive, metabolic chorioretinal dystrophy beginning in childhood.96 It is caused by a deficiency in the mitochondrial matrix enzyme ornithine aminotransferase (OATase), resulting in hyperornithinemia.97,98,99,100,101,102,103,104,105 The genetic mutation has been mapped to chromosome 10 (10q26).106
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


