Immunology of Uveitis
Hui Shao
Deming Sun
Shlomit Schaal
Henry J. Kaplan
Important advances have been made in understanding the immunopathologic basis of ocular diseases largely from developments in the field of immunology and molecular biology. Immunology has progressed from a simple dichotomy of Metchnikoff’s theory of cellular immunity and Ehrlich’s theory of antibody-mediated immunity to a better understanding of their complex relation in both generating and regulating the immune response. We are in an era in which many fundamental immunologic observations are being described not only at the cellular but also at the molecular level. These advances promise to provide an understanding of the pathogenesis of many inflammatory disorders and to direct the search for therapeutics. In this chapter, we discuss the generation and regulation of the immune response, present specific clinical applications in the context of the different types of hypersensitivity, and address additional immune mechanisms in uveitis.
Overview of the Immune Response
The immune response presumably evolved to protect the host from infectious pathogens, although with the increased longevity of our species it may have adapted to recognize and contain neoplastic cells. When a host encounters a protein or other immunogen that it recognizes as nonself (i.e., an antigen), a complex series of responses ensue that are designed to protect the host from possible harmful effects of the protein. In the initial phase, components of innate (natural) immunity intervene. Innate immunity (a) is present before the introduction of infectious microbes and other foreign macromolecules, (b) is not enhanced by prior exposure, and (c) protects the host immediately on contact with a pathogen.
While innate immunity is containing the antigen, adaptive (acquired) immunity is mounted to destroy the foreign substance. Adaptive immunity is stimulated by exposure to foreign substances, is extremely specific for particular macromolecules, and increases in magnitude with each successive exposure.1 Extensive interaction occurs between innate and adaptive immunity. Often, the type of adaptive immunity developed by a host—critical in successful clearance of a pathogen—is directly influenced by signals from the innate immune system.
Mammals sense pathogen invasion through pattern-recognition receptors, also called pattern-associated membrane proteins (PAMPs).103 A group of transmembrane proteins, Toll-like receptors (TLRs), play critical roles as pattern-recognition receptors. They are mainly expressed on antigen-presenting cells, such as macrophages or dendritic cells, and their signaling activates antigen-presenting cells to initiate the innate immune response. Ligand-receptor binding to each TLR results in both common TLR effects, such as inflammatory cytokine induction and/or upregulation of costimulatory molecule expression, as well as TLR-specific function, such as the induction of type I IFN. These immunoadjuvant effects are not only important for antimicrobial immunity, but are also involved in the development of autoimmunity.103
As our knowledge of the immune response increases, its complexity becomes more apparent. After generating an immune response to an antigen, the next important task is regulating that response. Although the immune response usually functions effectively, it occasionally goes awry and leads to autoimmunity.
Generation of the Immune Response
A complex series of events is initiated when the immune system responds to an antigen. The first line of defense is innate immunity, which is present before exposure to antigen. Components of this branch of immunity include physical barriers, such as skin and mucosal membranes; cells, such as neutrophils, eosinophils, basophils, macrophages and natural killer (NK) cells; and soluble plasma proteins, such as the complement system.
Neutrophils, the major cell population in acute inflammation, and tissue macrophages respond rapidly to chemotactic stimuli and subsequently phagocytose and destroy foreign particles without regard to antigen-specificity. Tissue mast cells and circulating basophils contain inflammation-inducing granules, the release of which is responsible for immediate hypersensitivity (allergic) reactions. Eosinophils, which also contain potent inflammatory granules, are known to function mainly in parasitic and helminthic infections. Additionally, the presence of eosinophils has been noted at sites of chronic allergy and inflammation. NK cells are large granular lymphocytes that kill target cells by osmotically lysing target cells, with exocytosed granules containing perforin and granzymes, and by inducing apoptosis (programmed cell death); this killing is not specific for any antigen and is categorized as part of innate immunity.1
While innate immunity initially “keeps the enemy at bay,” it also directs the adaptive immune response to deliver the definitive attack against the foreign invader. Furthermore, the adaptive immune response focuses and directs natural immune mechanisms to eliminate the antigen. The generation of acquired immunity entails (a) the production and secretion of antibody (immunoglobulin) into the blood and other body fluids (i.e., humoral immunity), and (b) the generation of sensitized lymphocytes (i.e., cellular immunity).
Humoral immunity involves soluble and circulatory factors in the immune system, such as antibodies and complement proteins. Whereas antibodies are generally considered part of adaptive immunity, the complement cascade participates in host defense by three different pathways. The classical pathway is activated by antigen–antibody complexes, the lectin pathway by pattern-recognition receptors (such as TLR), while the alternative pathway serves as an amplification mechanism for the complement cascade and can be activated by many different stimuli. Thus, the compement cascade is involved in both the innate and adaptive immune responses. Activation of the complement cascade leads to the generation of chemotactic molecules, cytotoxic complexes (i.e., the membrane attack complex) and covalent binding of complement proteins to the microbial cell surfaces. Complement activation on the surface of a microbial cell promotes adherence of the microbe to a phagocytic cell that is capable of phagocytosis and killing of the microbe. This process of “tagging” a cell with antibody or complement factors to facilitate phagocytosis is referred to as opsonization.
Cellular immunity is based on the function of the lymphocyte.2 Although lymphocytes are morphologically identical, they are functionally heterogeneous. They express an array of cell-surface molecules (markers), which define two major populations: T cells, B cells, and their subsets (Fig. 1).3
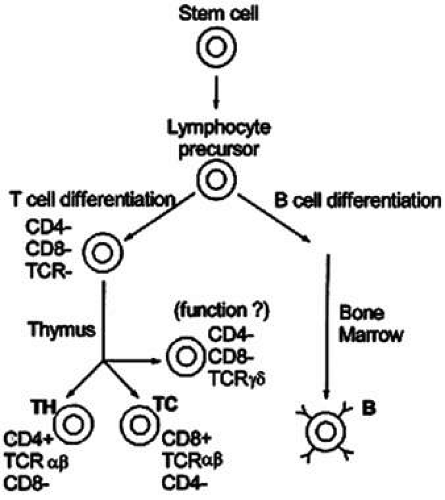 FIGURE 1. Lymphoid differentiation. Stem cells from the bone marrow migrate to the thymus, where hormonal factors stimulate differentiation into T cells. T cells subsequently differentiate further into αβ TCR CD4+, αβ TCR CD8+, and γδ TCR T cells. B lymphocytes are also derived from stem cells and differentiate in the bone marrow. Their cell surfaces express membrane-bound antibodies. TCR = T-cell receptor; Y = surface immunoglobulin; α, β, γ, δ = T-cell receptor subunits. |
Lymphocytes interact with antigen presenting cells (e.g. macrophages and/or dendritic cells), as well as each other, to allow selective expansion or suppression of specific populations (clones) of lymphocytes. Each lymphocyte clone has a unique receptor on its cell surface, which allows it to recognize and combine optimally with one antigen. Combination of this protein receptor with antigen results in the transmission of a signal across the cell membrane that activates the lymphocyte and results in selective proliferation and expansion of that lymphocyte clone (Fig. 2). Although numerous cellular interactions are involved in the generation of an immune response to antigen, our focus is on three distinct types: (a) antigen recognition by antigen-presenting cells (APC), such as macrophages and dendritic cells, and presentation to T cells (e.g. macrophage–T-cell interaction); (b) antibody production in response to antigen (T-cell–B-cell interaction); and (c) cytokine-mediated augmentation or inhibition of T-cell differentiation and proliferation (T-cell–T-cell interaction).
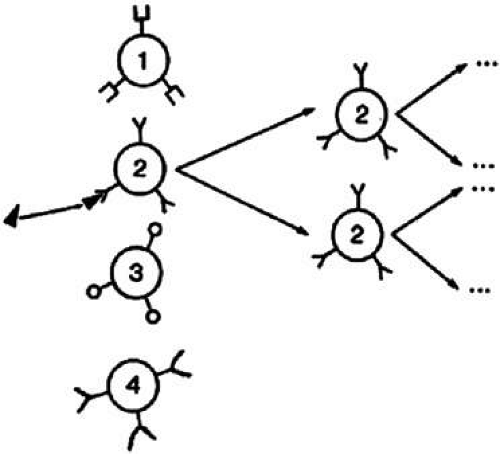 FIGURE 2. Theory of clonal expansion. The receptor on the cell surface of each lymphocyte clone is a unique protein that allows it to recognize and optimally combine with a specific antigen. After the combination of this receptor with the appropriate antigen, proliferation and expansion of that lymphocyte occur. Thus, the immune response can selectively focus its attention on the antigen or antigens being recognized. |
Antigen Presenting Cell (APC)–T-Cell Interaction
T-cell antigen recognition and activation depends on the presentation of the peptide antigen by specialized APCs, such as macrophages, B cells, and dendritic cells, which are accessory cells such as Langerhans cells in the skin or follicular dendritic cells in lymph nodes. All APCs digest proteins into peptide fragments and present these peptides within the clefts of special surface proteins coded by the major histocompatibility complex (MHC).
The MHC is a genetic segment on the short arm of chromosome 6 that codes for class I molecules (e.g., human leukocyte antigen (HLA)-A, HLA-B, and HLA-C) and class II molecules (HLA-DR, HLA-DP, HLA-DQ; Fig. 3) in addition to complement proteins and cytokines. Class I and II molecules were originally discovered as triggering T-cell responses leading to transplant rejection, hence the origin of the term histocompatibility.
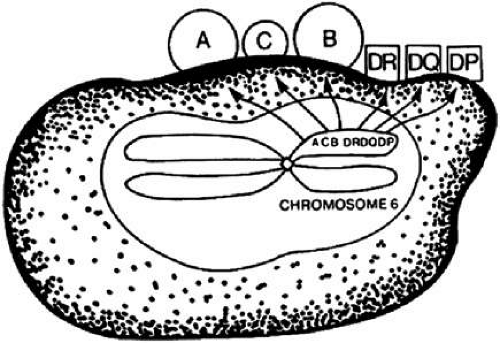 FIGURE 3. Major histocompatibility complex (MHC). Human chromosome 6 contains a genetic segment that codes for the class I MHC (HLA-A, HLA-B, and HLA-C) and class II MHC molecules (HLA-DR, HLA-DQ, and HLA-DP). These cell-surface molecules are glycoproteins present on many of the cells that are intimately involved in the recognition, activation, and effector phases of the immune response. HLA = human leukocyte antigen. |
The class II region, first identified in mice as the I region,4 codes for protein molecules expressed only on the cell membrane of macrophages, B cells, and dendritic cells. Class II-associated antigen presentation requires phagocytosis of extracellular antigens (e.g., bacterial proteins), followed by intracellular processing of the antigens. Activation of a major subset of T cells termed CD4+ helper T cells requires class II-associated antigen presentation; therefore, CD4+ T cells are called class II-restricted. (CD—cluster of differentiation—molecules are lymphocyte cell-surface markers; CD4+ designates helper T cells, and CD8+ identifies cytolytic T cells).
Class I molecules are expressed on almost all cells and are involved in presentation of endogenous antigens, such as tumor antigens, and viral proteins, which are not phagocytosed but enter the cell by viral infection. CD8+ cytolytic T lymphocytes (CTL) are class I-restricted and on activation destroy the entire host cell harboring the foreign antigen. The MHC molecule binds antigen within its clefts and the T-cell receptor (TCR) has regions that recognize the MHC complex and contact the foreign peptide (Fig. 4).5 The MHC-antigen complex must bind the antigen-specific TCR to achieve T-cell activation. Costimulatory molecules on APCs also bind with other T-cell membrane receptors during this APC-T-cell binding and are responsible for the development of either activation or tolerance to the specific antigen.1
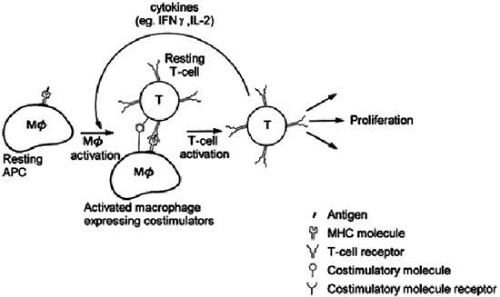 FIGURE 4. T-cell-macrophage interaction. T-cell activation requires recognition of antigen in conjunction with a class II MHC molecule, processed and presented by the macrophage. The antigen and class II major histocompatibility complex molecule are recognized by a specific T-cell receptor. Activated antigen-presenting cells (APC) express costimulatory molecules, which are required for T-cell activation. IL-2 is released by activated T cells, assisting in the subsequent clonal expansion and differentiation of T cells and also in the activation of resting APCs. IL-2 = interleukin-2. |
Antigen recognition is followed by clonal proliferation and expansion of those T lymphocytes that are directed at the involved antigen. Several antigen-specific memory T cells are also generated during this process, enabling the host to mount a greater and faster response on future exposure to the same antigen. Upon activation, the T cell elaborates many soluble factors called cytokines, including interleukin-2 (IL-2). IL-2 is the major T-cell growth factor, acting both in an autocrine and in a paracrine fashion, stimulating growth of not only other CD4+ and CD8+ T cells but also B cells and NK cells. The potent immunosuppressant drug cyclosporin inhibits the production of IL-2 by activated T cells, thereby blocking proliferation and differentiation of immune cells. Cytokines are not unique to T cells but are secreted by many different types of cells, enabling the immune system to orchestrate inflammatory responses.
In particular, interferon-gamma (IFN-γ) and tumor necrosis factor (TNF) secreted by T cells activate macrophages by setting off a cascade of upregulatory events, rendering the macrophage a more powerful and efficient killer of intracellular organisms. For example, on activation, the macrophage is induced to produce large amounts of nitric oxide, which is thought to inhibit bacterial and viral replication. While the proliferation of microorganisms is inhibited, potent proteases are released within the phagolysosomes, thereby destroying the pathogens more effectively. Also, angiogenic factors are released, such as vascular endothelial growth factor (VEGF) and fibroblast growth factor (FGF), which initiate endothelial cell migration and proliferation, and thus, tissue repair.
T-Cell–B-Cell Interaction
B-cell and helper T-cell cooperation entails activation and proliferation of both subsets of lymphocytes. As described earlier, T-cell activation requires antigen presentation, and B cells can serve well as APCs. After the antigen is recognized by a surface immunoglobulin on a B cell, the B cell is activated. The antigen is subsequently internalized, processed, and presented by the B cell in the context of a class II MHC molecule. The advantage of B cells over other APCs is that they are able to present antigen at a 104– to 106-fold lower concentration.1
Binding of a T cell to an antigen-presenting B cell leads to T-cell activation and release of IL-2, among other cytokines, promoting antigen-specific B-cell and T-cell proliferation. Binding of CD40, a B-cell surface receptor, to its complementary CD40 ligand on the activated T cell is also necessary for B-cell activation, differentiation, and survival. Other cytokines elaborated as a result of T-cell activation (e.g., IL-4, IL-5, transforming growth factor beta [TGF-β], and IFN-γ) serve to enhance antibody secretion and isotype switching.1
The production of antibody against most antigens requires this synergistic cooperation of helper T cells and B cells. The omission of either population results in limited or no antibody production. This is true in both the primary (i.e., on initial antigen exposure) and secondary (i.e., on subsequent antigen exposure) antibody responses.
B cells respond to a wider variety of antigens than T cells, including polysaccharides, lipids, as well as proteins. The selective activation of B cells is followed by differentiation into immunoglobulin secreting cells (i.e., plasma cells; Fig. 5). Memory B cells are also produced in this process of differentiation for a response of greater magnitude and speed on future encounter with the same antigen. Immunoglobulins are an important component of the response of the immune system to invasive pathogens. The basic structure of an immunoglobulin molecule consists of two heavy and two light chains, each composed of specific amino acid sequences (Fig. 6). There are five different classes (or isotypes) of immunoglobulin molecules based on the amino acid sequence in the constant region of their heavy chain (IgG, IgM, IgA, IgE, and IgD). The IgG molecule has the structure depicted in Figure 6. In contrast, the IgM molecule is a pentamer, consisting of five such units linked by a joining (J) chain (Fig. 7). The IgA molecule is most frequently a dimer, also joined by the J chain.
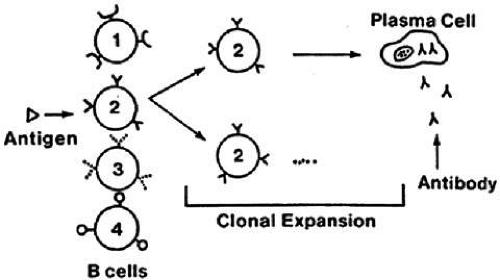 FIGURE 5. B-cell proliferation and differentiation. After B-cell activation by a specific antigen, clonal expansion occurs. The expanded population of B cells then differentiates into plasma cells, which secrete the immunoglobulin directed at the initially recognized antigen. |
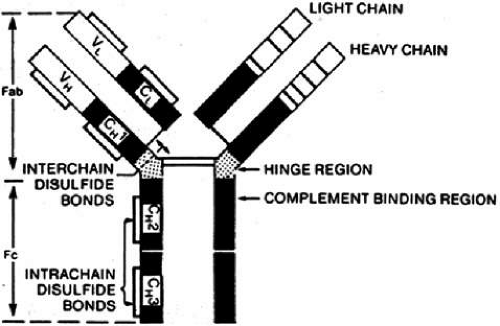 FIGURE 6. Immunoglobulin structure. The basic immunoglobulin molecule consists of two heavy and two light chains composed of specific amino acid sequences. The Fab (antigen-binding portion of the immunoglobulin molecule) contains the region of greatest variability in amino acid sequence (VL and VH, variable light- and heavy-chain domains) and a region of relatively constant amino acid sequence (CH1, constant heavy-chain 1; CL, constant light-chain domains). The Fc portion consists of only constant regions (CH2 and CH3) and contains the Fc receptor and complement-binding sites. |
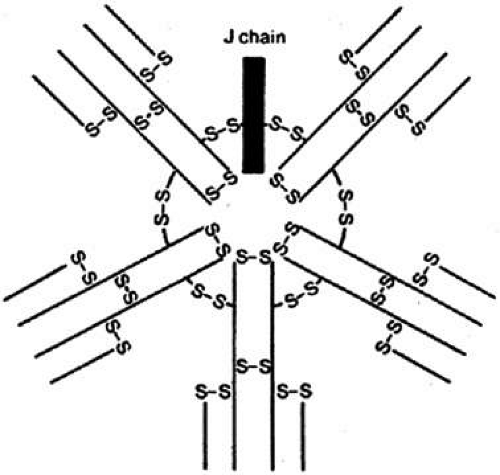 FIGURE 7. IgM. The IgM molecule is a pentamer consisting of five basic Ig units linked by disulfide bonds with a J chain. The biologic properties of the various immunoglobulin classes are distinctive and frequently determined by their physical properties. The IgM molecule does not cross the placenta and, therefore, is helpful in the diagnosis of congenital ocular toxoplasmosis. |
The isotype of antibody that is produced can be important diagnostically. After initial contact with antigen, the immune response produces antibody in what is called the primary response (Fig. 8). The first class of antibody produced is IgM. Only a low titer of IgM antibody is produced, however, and it rapidly declines to undetectable serum levels. During the second week of the immune response, an IgM to an IgA, IgE, or IgG isotype switch occurs, depending on the cytokine produced by the helper T cell, and high titers of the new isotype antibody are produced in the plasma. These titers persist long after the onset of the immune response. When the immune response again encounters the same antigen, a secondary antibody response occurs. The main antibody produced by the memory B cells is IgA, IgE, or IgG, which reaches even higher serum levels and persists even longer. In contrast, IgM antibody titers appear transiently, remain much lower, and rapidly return to undetectable levels.
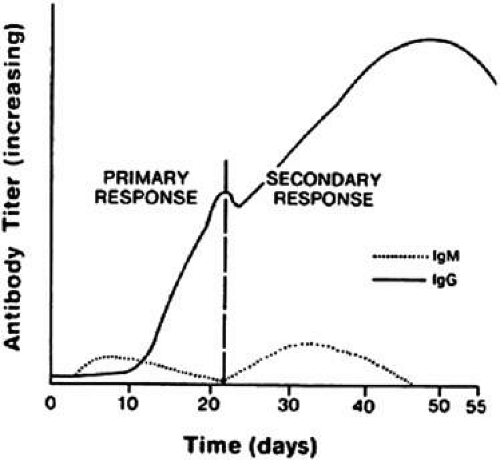 FIGURE 8. Primary and secondary antibody responses. In this example, the primary antibody response to an antigen is characterized by a switch from IgM to IgG. Subsequent reexposure (i.e., the secondary antibody response) to this antigen preferentially produces IgG antibody. |
T-Cell–T-Cell Interaction
The interplay between subsets of T cells is achieved through secretion of cytokines, which modulate the immune response. For example, the immune response to antigen can result in the generation of CD8+ cytotoxic T lymphocytes (CTLs) with the ability to kill virus-infected host cells or allogeneic transplanted cells (i.e., transplanted cells from genetically different individuals of the same species). Optimal generation of CTLs from their precursors (pre-CTLs) requires cytokine release from helper T cells.
Initially, pre-CTLs bind peptide antigens expressed in the context of class I MHC molecules on the surface of target cells, and are thus, activated. Additional ligand-receptor interactions between pre-CTLs and target cells, in the presence of the cytokines IL-2 and IFN-γ, subsequently lead to full activation and differentiation of pre-CTLs to CTLs capable of killing target cells. IL-2 and IFN-γ, both of which are necessary for the full activation and differentiation of CTLs, are produced mainly by neighboring helper T cells, which are concurrently activated in response to viral infection or an allogeneic graft. Helper T cells, however, are activated by a different peptide sequence that binds to class II MHC molecules on APCs. Dendritic cells especially have been implicated in presenting foreign proteins expressed by viruses or allogeneic grafts by endocytosing fragments of apoptotic cells that express these proteins. Therefore, an intimate cooperation between class I-restricted CD8+ T cells (i.e., pre-CTLs) and class II-restricted CD4+ helper T cells leads to the development of CTLs capable of recognizing and killing target cells that express foreign proteins.
This accessory ligand-receptor conjugation between the target cell and the antigen-specific CTL is required for efficient killing. After this conjugate is formed, CTLs deliver their lethal hit mainly by granule exocytosis (e.g., perforin, a membrane pore-forming protein; and granzymes, enzymes containing reactive serines at their active site), leading to apoptosis and osmotic lysis.
Many apoptotic mechanisms in the immune system have been linked to the interaction of Fas ligand, a cell-surface protein on the killer cell, with its receptor Fas (also known as CD95) on the target cell. The binding of this receptor to its ligand triggers a cascade of intracellular protein–protein interactions and proteolytic activities, culminating in apoptosis of the target cell. This pathway is thought to be more important in immune regulation—that is, for controlling excessive lymphocyte activation especially against self-antigens—than for CTL functions.1,6
Regulation of the Immune Response
Because multiple cell–cell interactions occur during the generation of an immune response, it is necessary to regulate these interactions so that the net effect is appropriate, minimizing the inadvertent destruction of innocent host bystander cells.
Multiple homeostatic controls exist to achieve immunoregulation, of which we briefly describe the principal mechanisms. So far, clonal expansion has been considered in the context of selection by antigen. As such, the regulation of clonal expansion can be achieved by antigen removal by secreted antibody or T cells. The depletion of antigen for lymphocyte receptors serves as negative feedback for antigen-driven clonal expansion. Thus, only memory cells remain, which require subsequent activation by antigen for regeneration of a full response.
Immunologic tolerance is an important mechanism for immune regulation, applicable to both B and T lymphocytes. Studies have shown that tolerance is antigen-specific, and thus, likely achieved by either deletion or inactivation of specific B- or T-cell clones (i.e., clonal deletion or clonal anergy). Immature lymphocytes are more susceptible to induction of tolerance than mature, functional cells. In the thymus, immature T cells are thought to be exposed to self-antigens; if a T cell recognizes any self-antigen, it is eliminated by apoptosis. This mechanism of tolerance is referred to as clonal deletion because it involves cell death. The purpose of this central tolerance (central because it occurs in the thymus) is the prevention of autoimmunity.1
Under special conditions, tolerance may be induced in mature lymphocytes, away from the central lymphoid tissues (peripheral tolerance). For example, a large concentration of foreign polysaccharides injected intravenously does not stimulate a B-cell response; even subsequent exposure to immunogenic doses of the antigen fails to activate B cells. Peripheral CD4+ T-cell tolerance has also been achieved in other ways, including high-dose intravenous administration of proteins without adjuvant (high-dose tolerance), repeated low-dose exposure to proteins without adjuvant (low-dose tolerance), and oral administration of proteins (oral tolerance). T-cell tolerance is generally induced at lower concentrations of protein than is B-cell tolerance and lasts longer. Moreover, without CD4+ T-cell help, the B-cell response to low levels of protein antigen is less. The mechanisms involved in peripheral tolerance are clonal anergy as well as clonal deletion—the former referring to functional inactivation without cell death.1
Because the antigen receptor on lymphocyte cell membranes is the only clonally distributed structure, it is likely involved in cell regulation. This can occur through either cell–receptor interaction with antigen or with factors directed at the antigen receptor itself (i.e., idiotype–anti-idiotype recognition). The precise details of the idiotypic–anti-idiotypic networks of immunoregulation are not discussed here because it has a more theoretic basis with insufficient experimental evidence. Regulation of the immune response is also maintained through the function of CD4+ and CD8+ T cell subsets, referred to as regulatory T cells (see Ocular Immune Privilege).104
Different types of immune responses may be suppressed. CD4+ T cells and these cells have been subclassified as T helper 1 (TH1) and T helper 2 (TH2) cells. TH1 cells direct the immune reaction toward a cell-mediated response through expression of IFN-γ, IL-2, and TNF-α; TH2 cells elaborate IL-4, IL-5, and IL-10, leading to IgE-mediated humoral immunity. Interestingly, IFN-γ inhibits the IL-4-mediated B-cell isotype switch to IgE; conversely, IL-4 inhibits IFN-γ-mediated macrophage activation. The trigger for the TH1 response has been shown to be IL-12 secreted by the activated macrophage (Fig. 9).1
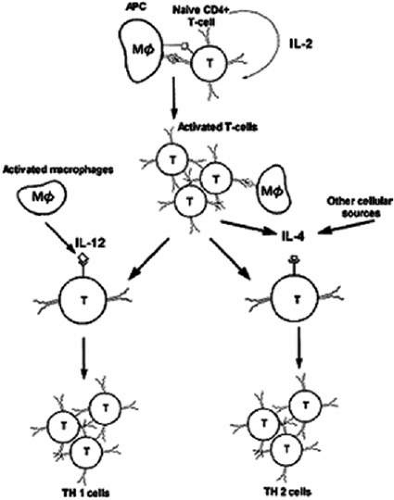 FIGURE 9. Induction of TH1 and TH2 cells. Cytokines produced by the innate immune response to pathogens or during early phases of specific immunity can influence the differentiation of naive CD4+ T cells into TH1 or TH2 cells. IL-12, made by activated macrophages or dendritic cells, is a strong inducer of TH1-cell development. IL-4, which may be secreted during the initial activation of naive T cells, favors induction of TH2 cells. (Adapted from Abbas AK, Lichtman AH, Pober JS: Cellular and Molecular Immunology, 3rd ed. Philadelphia, WB Saunders, 1997:272.) |
In certain conditions, however, the desirable immune response becomes suppressed, leading to a detrimental outcome. For example, a TH2 response in leprosy is associated with chronicity and greater host-tissue destruction, whereas a TH1 response theoretically would be more effective. One explanation for the tendency toward a TH2 response may be as follows. IL-4 is initially produced in small amounts by the T cell at the time of its initial activation, and its concentration increases when high concentrations of antigen persist. If this is combined with the absence of IL-12 secretion, a TH2 response will predominate. Genetic factors have also been implicated in the predominance of one type of response over the other.1
Ocular Immune Privilege
The eye is considered to be an immunologically privileged organ. This allows for the existence of immune defense mechanisms that protect the eye from infectious pathogens, while simultaneously controlling the severity of intraocular inflammation to preserve organ function and integrity. Experimental evidence for ocular immune privilege has been reported in several separate studies7,8,9 showing that tissue grafts or tumor cells placed in the anterior chamber are not rejected, whereas they are rejected rapidly when placed elsewhere, such as in subcutaneous tissue. The high success rate of allogeneic orthotopic corneal grafts in mouse and human eyes, even in the presence of major histocompatibility differences, is another example of ocular immune privilege.10
Features that confer immune privilege in the eye include a blood–tissue barrier, restricted lymphatic drainage, intraocular fluid drainage into the circulatory system, and reduced expression of MHC class I, II and costimulatory molecules on parenchymal cells.11 Other mechanisms exist that contribute to the control of the immune response within the eye:
Complement inhibition. Several complement regulatory proteins, including membrane cofactor protein (MCP), decay-accelerating factor (DAF), and CD59, have been identified in ocular tissue. The putative role of these molecules is protection of intraocular tissues from complement-mediated destruction.12
Clonal deletion. Fas ligand is constitutively expressed in the iris, ciliary body, cornea, and retina. Evidence from tissue grafts in vivo and tissue cultures in vitro support the view that constitutive expression of Fas ligand in a tissue leads to deletion of the Fas+ T cells that enter that tissue and recognize antigen.13
Clonal anergy. Aqueous humor contains many immunomodulatory substances, including TGF-β2 (a powerful inhibitor of T-cell and B-cell proliferation), free cortisol, and IL-1 receptor antagonist. Cultures of primed T cells that encounter antigen in the presence of aqueous humor failed to undergo proliferation, failed to produce cytokines, such as IFN-γ, and actually inhibited other primed T cells from producing IFN-γ. When soluble TGF-β2 receptor was used to “quench” the TGF-β2 in the culture medium, IFN-γ production by the primed T cells was restored.14
Immune deviation. Investigations by Kaplan, Neiderkorn and Streilein7,116 demonstrated that histoincompatible lymphoid or tumor cells placed in the anterior chamber evoke a deviant systemic immune response characterized by suppressed delayed-type hypersensitivity, enhanced antibody production and inhibition of cell-mediated cytotoxicity. Mice injected with allogeneic tumor cells into the anterior chamber lost the ability to reject skin grafts from the allogeneic donor; delayed-type hypersensitivity reactions against allogeneic tissue were abrogated. This phenomenon is termed anterior chamber-associated immune deviation (ACAID).
In subsequent studies, foreign protein injection into the anterior chamber revealed a predilection of the ocular immune system toward a TH2 or humoral immune response. A potentially important application of this phenomenon was demonstrated in the setting of experimental autoimmune uveoretinitis (EAU), in which injection of the autoantigen into the anterior chamber before the uveitogenic regimen inhibited uveitis.14
Role of Regulatory T Cells. Spleens of mice in which ACAID has been induced contain T cells that are capable of conferring ACAID when adoptively transferred into naive recipients, regulatory T cells. Thus, similar to the donors, these naive recipients become unable to mount a delayed-type hypersensitivity response to the antigen.15 Experimental evidence supports the view that either a soluble signal from the eye or antigen-bound APCs from the anterior chamber enter the bloodstream and migrate to the spleen. In the spleen, ACAID-mediating T cells (CD8+ cells)–regulatory T cells–are generated. Some of the responding CD8+ T cells secrete TGF-β, further suppressing CD4+ T-cell activation, both locally and eventually systemically.14 The active role of both the eye and the spleen in ACAID is highlighted by the finding that removal of the spleen or the eye within the first 3 days after exposure to antigen results in the typical immune response.16,17 It is apparent that several cell sub-types “cross-talk” within the spleen to promote the induction of ACAID.117
Role of NKT Cells. CD1-reactive NKT cells are required for the development of ACAID. For example, CD1 knockout mice were unable to develop ACAID unless they were reconstituted with NKT cells together with CD1(+) APC; specific antibody depletion of NKT cells in vivo abrogated the development of ACAID; anti-CD1 monoclonal antibody treatment of wild-type mice prevented ACAID development. Significantly, CD1-reactive NKT cells were not required for intravenously induced systemic tolerance, thereby establishing that different mechanisms mediate development of tolerance to antigens inoculated by these routes. A critical role for NKT cells in ACAID suggests a defect in NKT cells may be involved in the development of autoimmunity.105
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


