The classic risk factors for the development of primary open angle glaucoma (POAG) are age, race, family history, and elevated intraocular pressure (IOP). More recently, corneal thickness has also been identified as a risk factor for glaucoma.1 The role of heredity in POAG has long been supported by a range of investigations including epidemiological studies, twins studies, reports of large families in which glaucoma is inherited as a Mendelian trait, and studies with animal models of glaucoma.2,3,4,5,6,7,8,9,10,11,12,13,14,15,16,17,18,19 In the last 15 years, several genes have been identified that cause or contribute risk for developing glaucoma. The specific defects in the genetic code that cause approximately 5% of POAG cases have been discovered, and many genetic factors have been detected that contribute risk for development of more complex genetic forms of glaucoma. While some of the identified glaucoma genes (i.e., optineurin, TANK binding kinase, and toll-like receptor 4) have been connected to one another within a common biological pathway, most of the genes and risk factors for glaucoma do not have obvious relationships. Recent discoveries have confirmed that glaucoma has a strong genetic basis. However, many more glaucoma genes and risk factors remain to be identified, and additional functional studies of these genes will be necessary to clarify the molecular mechanisms of inherited glaucoma and to facilitate assessment of genetic risk for disease.
This chapter will focus on the genetic basis of POAG. Great advances have been made in genetic studies of other forms of glaucoma, including secondary glaucomas, primary congenital glaucoma, and primary angle closure glaucoma, which have been reviewed elsewhere.20,21,22,23,24,25,26,27
SIGNIFICANCE
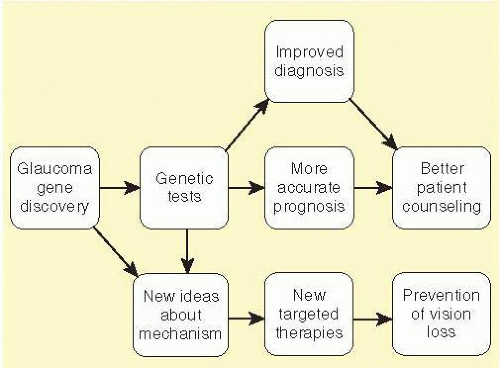
POAG is a common cause of visual disability in millions of Americans and disproportionately affects those of African heritage. The specific causes of POAG are poorly understood at the molecular level, which is a major obstacle to early diagnosis and the development of new therapies for this condition. The discovery of POAG genes will ultimately provide many benefits to the research and medical communities (Fig. 50.1). The identity of POAG genes may facilitate the development of genetic tests to accurately identify those patients at greatest risk for developing glaucoma so that treatments may be initiated before any vision is lost. Moreover, correlations between genetic defects and associated clinical course (genotype-phenotype associations) have the potential to arm physicians with better prognostic data for the care and education of their patients. A patient’s genome may eventually inform physicians not only about which diseases they are most likely to develop, but also which therapies are most likely to be beneficial. Moreover, as the specific causes of POAG are discovered at the molecular genetic level, it will likely be possible to target these defects more specifically, either with conventional medical or surgical treatments or with gene therapies designed to more directly correct pathogenic defects.
FIG. 50.1 Potential benefits from studies to identify glaucoma-causing genes.
MENDELIAN (SINGLE GENE) VERSUS COMPLEX (MANY GENE) DISEASE
The heredity of POAG is complex due to the actions of many genes. Some cases of glaucoma have a relatively simple genetic basis (i.e., caused primarily by the action of a single gene). Large pedigree-based studies have identified such genes (myocilin,28 optineurin,29 WD domain 36,30 TANK binding kinase 1,31 and ankyrin and SOCS box 1032). These genes have each been reported to cause POAG with little influence from other genes or the environment. Such forms of glaucoma are transmitted through families with simple inheritance patterns (usually autosomal dominant) akin to the transmission patterns described by Mendel. As a result, single gene forms of disease are often said to have Mendelian inheritance patterns. Although the evidence for some reported genes is stronger than others, individuals that inherit a mutation in a Mendelian glaucoma gene almost always develop glaucoma, and such mutations are almost never seen in normal individuals.
Together, mutations in the known Mendelian glaucoma genes are responsible for approximately 5% of POAG cases. For example, glaucoma-causing mutations in the myocilin (MYOC) gene were shown to be responsible for approximately 4% of cases of POAG and are usually characterized by markedly elevated IOP.28,33 Mutations in another gene optineurin (OPTN) have been associated with 1% to 2% of cases of normal tension glaucoma (NTG).29,34,35 The protein encoded by OPTN has a key function in autophagy,36 a process by which accumulating protein or defective organelles are digested into basic building blocks, which can be recycled within the cell.37 Another gene that is involved in autophagy, TANK binding kinase 1 (TBK1), has also been associated with NTG.31 The role of additional genes (WDR36, NTF4, and ASB10) in glaucoma pathogenesis is less clear.30,32,38,39,40,41,42,43 The known Mendelian genes are described in more detail in the next section.
Population-based studies of complex genetic forms of glaucoma (i.e., caused primarily by the combined actions of several genes) have identified several genes that each contribute small risk for POAG. These genetic factors are commonly observed in patients and controls, and each contributes small risk for disease. There appear to be many such POAG genetic “risk factors” (and POAG environmental factors), most of which have not yet been identified. Due to the large number of independently inherited factors that together promote disease, this form of POAG may not have a clear inheritance pattern, but may be enriched within families or ethnic populations. Such risk factors are frequently seen in normal subjects, but are statistically more common among POAG patients, and each of these genes or POAG risk factors is incapable of causing disease on its own. For example, a genetic variation in the CDKN2B-AS1 gene (rs4656461) has been detected in 17% of a large cohort of POAG patients and 12% of matched controls (P = 6.0 × 10-14). These data suggests that variants in the CDKN2B-AS1 gene may be associated with increased risk for glaucoma (odds ratio = 1.51), but do not cause disease on their own.44
MENDELIAN (SINGLE GENE) DISEASE GENES
The first Mendelian glaucoma gene was mapped to a region of chromosome 1q designated GLC1A in 1997. The locus name GLC1A is an acronym in which “GLC” indicates the presence of an open angle glaucoma gene, the “1” indicates adult onset, and the loci are numbered in order of their discovery by the final letter. As of 2012, a total of 16 (GLC1A-GLC1P) loci have been mapped (Table 50.1), and the glaucoma genes have been reported at 6 of these loci and are described in more detail below.
Myocilin (MYOC)—JOAG and POAG
Family-based studies of juvenile-onset open angle glaucoma (JOAG) mapped the first glaucoma gene to the chromosome 1q locus (GLC1A).45 Later, JOAG patients were found to have glaucoma-causing mutations in one of the genes in the GLC1A locus.28 This gene, myocilin (MYOC), has unknown function and shares little instructive homology with other known genes.
Myocilin Mutations Correlate with Glaucoma Features
Many myocilin mutations have been detected, some of which are associated with glaucoma and some of which are benign.33,46,47 Two classes of myocilin mutations have been identified. One set of mutations are associated with JOAG.33,46 Individuals with these mutations develop early onset of disease, markedly elevated IOP, and strong autosomal dominant inheritance.48 JOAG patients with myocilin mutations typically fail to achieve adequate IOP control with topical medications and require surgical intervention.49 A second set of myocilin mutations are associated with adult-onset POAG.33,46 Individuals with these mutations respond to topical medications and require surgical intervention at the same frequency as do POAG patients that have no myocilin mutations.50 One myocilin mutation, Gln368Stop, is associated with approximately 1% of adult-onset POAG cases in Caucasians and African Americans from the United States, Canada, Australia, and Europe, but is not observed in Asian POAG patients.33,46 The singular feature of myocilin-related glaucoma is markedly high IOP especially with JOAG-related glaucoma. Furthermore, the magnitude of maximum IOP is strongly correlated with particular myocilin mutations.48
TABLE 50-1 Identified Chromosomal Loci and Genes That Cause Mendelian (Single-Gene) Forms of POAG
Locus Name
Chromosomal Location
Gene at the Locus
Type of Glaucoma
Fraction of POAG Cases
Genetic Study Evidence
Cell Culture, Organ Culture, or Animal Models of Disease
GLC1A
1q23-q25
Myocilin (MYOC)
JOAG,
POAG
3%-5% POAG
Strong (multiple reports)
Organ culture and transgenic mice
GLC1B
2cen-q13
Unknown
POAG
—
Unconfirmed
—
GLC1C
3q21-q24
Unknown
POAG
—
Strong (multiple reports)
—
GLC1D
8q23
Unknown
POAG
—
Unconfirmed
—
GLC1E
10p13
Optineurin (OPTN)
NTG
1%-2% NTG
Moderate (unconfirmed)
Transgenic mice
GLC1F
7q35-q36
Ankyrin and SOCS box 10 (ASB10)
POAG
—
Conflicting data
Anterior segment organ culture
GLC1G
5q22
WD repeat domain 36 (WDR36)
POAG
—
Conflicting data
Cell culture
GLC1H
2p16-p15
Unknown
POAG
—
Unconfirmed
—
GLC1I
15q11-q13
Unknown
POAG
—
Unconfirmed
—
GLC1J
9q22
Unknown
JOAG
—
Unconfirmed
—
GLC1K
20p12
Unknown
JOAG
—
Unconfirmed
—
GLC1L
3p22-p21
Unknown
NTG
—
Unconfirmed
—
GLC1M
5q22.1-q32
Unknown
JOAG
—
Unconfirmed
—
GLC1N
15q
Unknown
JOAG
—
Unconfirmed
—
GLC1O
19q13.33
Neurotrophin 4 (NTF4)
POAG
—
Conflicting data
—
GLC1P
12q14
TANK binding kinase 1 (TBK1)
NTG
0.4%-1.3% NTG
Moderate (two reports)
Cell culture
Prevalence of Myocilin Mutations in POAG
Mutations in myocilin are responsible for 4% to 60% of JOAG cases and 3% to 4% of cases of adult-onset POAG.51,52 As a result, myocilin-related glaucoma is currently the most common known cause of glaucoma worldwide.
Mechanism of Disease
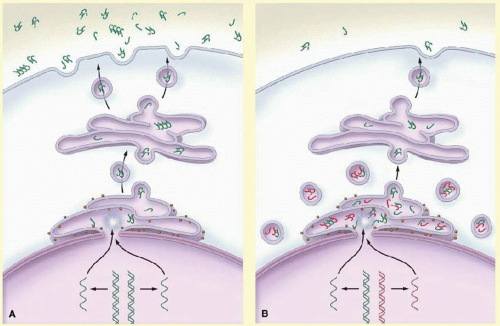
The normal function of myocilin is unknown; however, the wild-type protein is produced in many tissues of the eye, including the trabecular meshwork where it is secreted into the aqueous humor.53,54,55 Mutant myocilin protein is retained within the trabecular meshwork which leads to an intracellular accumulation of abnormal protein.55,56 In vitro studies and research with transgenic mice have shown that defective myocilin protein may stimulate the unfolded protein response as a mechanism to clear collections of abnormal myocilin protein.57,58 Intracellular accumulation of myocilin protein may subsequently lead to trabecular meshwork cell death, reduced facility of outflow, elevated IOP, retinal ganglion cell death, and ultimately to glaucoma (Fig. 50.2).57,58,59,60 These investigations have suggested novel therapies for myocilin-related glaucoma. Treatment with drugts that inhibit the unfolded protein response, sodium 4-phenylbuturate (PBA), prevent elevated IOP and glaucoma in mice.57,58,61 PBA has promise as a therapy for human glaucoma, but remains to be tested.
Genetic Testing and Gene-Directed Therapies
Genetic testing for myocilin is widely available and can be arranged via many laboratories (www.genetests.org). Genetic testing under the guidance of experienced ophthalmologists and/or genetic counselors may be warranted for patients with JOAG, given the high prevalence of myocilin mutations in such patients, especially when there is a strong family history of disease and markedly elevated IOP. Similarly, there may be utility in testing for myocilin mutations in patients with POAG that have features of JOAG, such as relatively early onset of disease, strong family history, and markedly elevated IOP. Glaucoma patients that have relatives with proven myocilin-related glaucoma are also at high risk and may benefit from testing. Given the relatively low prevalence of myocilin mutations in unselected glaucoma patients, however, widespread mutation testing is not currently indicated.35
FIG. 50.2 Proposed pathways for normal secretion of myocilin into the aqueous humor and for secretion reduced by a MYOC mutation. A: Wild-type myocilin protein (green symbols) is produced in the endothelial cells of the trabecular meshwork and passes through the secretory pathway to reach the extracellular space. In the first step in this process, messenger RNA (mRNA) is transcribed from the gene encoding myocilin (MYOC) and is delivered to ribosomes at the endoplasmic reticulum, where the mRNA directs the synthesis of myocilin. Next, transport vesicles convey myocilin to the cell membrane through the Golgi apparatus. These vesicles fuse with the cell membrane and release myocilin into the extracellular space and aqueous humor. Along the secretory pathway, molecules of myocilin may associate with each other and form multimers (dimers and tetramers are depicted). B: Heterozygous mutations of the MYOC gene are associated with an autosomal dominant form of glaucoma. The wild-type copy of MYOC encodes normal myocilin protein (green symbols), and the mutant MYOC copy encodes mutant myocilin protein (red symbols). Myocilin protein forms multimers that may be composed of both wild-type and mutant subunits. Secretion of mutant myocilin protein and multimers containing mutant subunits is greatly reduced, leading to the retention of the mutant protein in the endoplasmic reticulum and intracellular vesicles of trabecular-meshwork cells. (From Kwon YH, Fingert JH, Kuehn MH, et al. Primary open-angle glaucoma. N Engl J Med. 2009;360:1113-1124.)
Optineurin (OPTN)—POAG/NTG
Studies of a large normal tension glaucoma (NTG) pedigree mapped a glaucoma gene to the chromosome 10 (GLC1E) locus.62 Glaucoma-causing mutations have subsequently been discovered in the optineurin (OPTN) gene, demonstrating that it is the glaucoma gene in the chromosome 10 GLC1E locus.29
Prevalence of OPTN Mutations in POAG
Initial reports suggested that as much as 17% of POAG may be caused by mutations in OPTN29; however, most subsequent studies have indicated that OPTN is responsible for a small proportion of POAG cases (<1%)34,63,64 and 1% to 3% of NTG.34,63,64 One OPTN mutation, Glu50Lys, has been strongly associated with glaucoma in many research reports and accounts for the majority of optineurinrelated disease,29,34 while there are mixed data about another mutation, Met98Lys, which appears to have a more important role in glaucoma in Asian populations than it does in Caucasian subjects.34,65
Mechanism of Disease
The biological pathway that leads from mutations in OPTN to the development of NTG has been explored using transgenic animals. Mice engineered to carry the Glu-50Lys OPTN mutation have been shown to develop signs of glaucoma, retinal ganglion cell loss, and axon loss in the absence of elevated IOP.66 Studies of these transgenic OPTN mice have confirmed the pathogenicity of the Glu-50Lys mutation and may provide future insights into the pathogenesis of glaucoma.
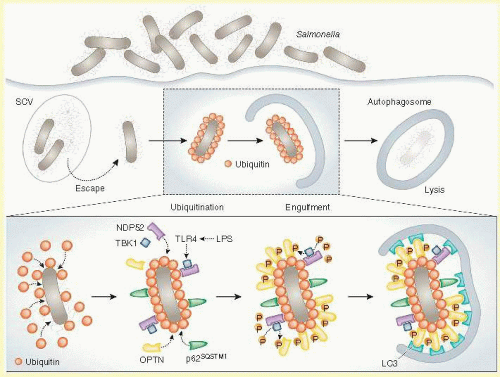
FIG. 50.3 TBK1-phosphorylated optineurin targets intracellular bacteria to xenophagic degradation. Upon infection, most Salmonella are sequestered into Salmonella-containing vacuoles (SCVs), but some can escape from SCVs and proliferate in the cytosol. To prevent this, cytosolic bacteria are rapidly ubiquitinated, leading to the recruitment of several autophagic receptors including NDP52 and p62SQSTM1 as well as that of the TANK-binding kinase 1 (TBK1). Until recently, the mechanism by which TBK1 would limit bacterial proliferation was not fully understood. Now, it has been discovered that, in response to bacterial products such as lipopolysaccharide (LPS), the pattern recognition receptor Toll-like receptor 4 (TLR4) activates TBK1, leading to the phosphorylation of another autophagic receptor, optineurin (OPTN), on Ser177. Phosphorylated OPTN has a high affinity for the autophagic protein LC3, thereby guiding (together with NDP52 and p62SQSTM1) ubiquitinated bacteria to the autophagic machinery and allowing for their elimination by xenophagy. (Reprinted by permission from Galluzzi L, Kepp O, Kroemer G. Autophagy and innate immunity ally against bacterial invasion. EMBO J. 2011;30:3213-3214.)
Recent studies of innate immunity have shown that OPTN has a central role in autophagy.36,67 When intracellular bacteria are recognized as pathogens, they are cleared from infected cells by the cellular process autophagy (Fig. 50.3), that includes (1) marking of bacteria for elimination with ubiquitin tags and (2) stimulation of a signaling pathway that includes molecules produced by other NTG genes, toll-like receptor 4 (TLR4), and TBK1. When the receptor TLR4 recognizes bacterial antigens such as lipopolysaccharide it activates the kinase TBK1, which subsequently phosphorylates and activates OPTN. Activated OPTN then binds to ubiquinated bacteria and brings in key components of the autophagosome (LC3, NDP52, and SQSTM1) that promote digestion of the pathogens.36,67 While there is little evidence to suggest that optinuerin-related glaucoma is incited by bacterial pathogens, similar processes have been implicated in clearance of accumulated abnormal intracellular proteins and defective organelles.37 Three known NTG genes, OPTN, TBK1, and TLR4, have important roles in autophagy, which suggests that mutations in these genes may cause glaucoma via dysregulation of autophagy. The connection between autophagy and apoptosis of retinal ganglion cells in glaucoma, however, is unclear. Some studies have suggested that upregulation of autophagy may promote apoptosis,68 while others have suggested that in some circumstances autophagy may have some protective effects and inhibit apoptosis.69,70,71
Only gold members can continue reading. Log In or Register to continue