Chapter52 Hereditary optic neuropathies
The hereditary optic neuropathies comprise a group of disorders in which the cause of optic nerve dysfunction appears to be hereditary, based on familial expression or genetic analysis. Clinical variability, both within and among families with the same disease, often makes recognition and classification difficult.1–3 Inherited optic neuropathies are often classified by pattern of transmission, most commonly autosomal dominant, autosomal recessive, and maternal (via mitochondrial DNA (mtDNA)). The same genetic defect may not be responsible for all pedigrees with optic neuropathy inherited in a similar fashion. Similarly, different genetic defects may cause identical or similar phenotypes – some inherited in the same manner, others not. Alternatively, the same genetic defect may result in different clinical expression, although the pattern of inheritance should be consistent. Also, single cases are often presumed or proven to be caused by inherited genetic defects, making the pattern of familial transmission unavailable as an aid to classification.
The inherited optic neuropathies typically manifest as symmetric, bilateral, painless, central visual loss. In many of these disorders, the papillomacular nerve fiber bundle is affected, with resultant central or cecocentral scotomas. The exact location of initial pathology along the ganglion cell and its axon, and the pathophysiologic mechanisms of optic nerve injury remain unknown, but mitochondrial dysfunction plays a central role in the pathogenesis of most, if not all, of the inherited optic neuropathies.2,3 Optic nerve damage is usually permanent and may be progressive. Once optic atrophy is observed, substantial nerve injury has already occurred.
In some of the hereditary optic neuropathies, optic nerve dysfunction is isolated. In others, various neurologic and systemic abnormalities are regularly observed and inherited diseases with primarily neurologic or systemic manifestations, such as the multisystem degenerations, can include optic atrophy. In this chapter the hereditary optic neuropathies are a classified into three major groups:1
1. Those that occur primarily without associated neurologic or systemic signs.
2. Those that frequently have associated neurologic or systemic signs.
3. Those in which the optic neuropathy is usually recognized as secondary in the overall disease process.
Monosymptomatic hereditary optic neuropathies
Leber’s hereditary optic neuropathy
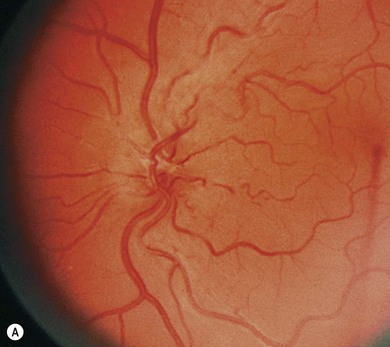
Leber’s hereditary optic neuropathy (LHON) is one of the first diseases to be etiologically linked to specific mtDNA defects.1–3 It has a minimum point prevalence of 1 in 31 000 in the northeast of England, 1 in 39 000 in The Netherlands and 1 in 50 000 in Finland.3 Age of onset typically occurs between 15 and 35, but may range from 1 to 87 years. LHON is expressed predominantly in males of the lineage, a feature not expected from mtDNA inheritance. Between 25% and 50% of male and 5% and 10% of female LHON carriers will have vision loss during their lifetimes. Visual loss is bilateral, painless, central, and usually abrupt in onset. In about 50% of cases, the vision loss is recognized as occurring sequentially, with one eye affected weeks to months before second eye involvement. Within 1 year, both eyes are invariably affected. Vision worsens in each eye over weeks or months, and usually deteriorates to acuities of 20/200 or worse. Color vision is affected early and severely, and visual fields typically show central or cecocentral defects. Funduscopic abnormalities may be seen in patients with LHON and in their asymptomatic maternal relatives. Especially during the acute phase of visual loss, there may be hyperemia of the optic nerve head, dilation and tortuosity of vessels, hemorrhages, circumpapillary telangiectatic microangiopathy, or circumpapillary nerve fiber layer swelling (pseudoedema) (Fig. 52.1). Eventually, the only fundus findings will be optic atrophy with nerve fiber layer dropout, especially in the papillomacular bundle. There may be non-glaucomatous cupping of the disc and arterial attenuation. In most LHON patients, visual loss is the only manifestation of the disease. Some pedigrees have family members with associated cardiac conduction abnormalities, especially pre-excitation syndromes. Minor neurologic and skeletal abnormalities have been reported in some patients, as has multiple sclerosis-like disease.
LHON pedigrees follow a maternal inheritance pattern, and the disease has been linked to point mutations in the mtDNA1–4 (Fig. 52.2). Three point mutations in the mtDNA, known as the “primary LHON mutations,” cause about 90% of cases of LHON worldwide. They are located at mtDNA nucleotide positions 11778 (69% of cases), 3460 (13% of cases), and 14484 (14% of cases) (see Fig. 52.2). Other point mutations have been found with a greater frequency in LHON patients than controls, but caution must be taken in assuming a causal significance for these secondary mutations. Some may truly be primary mutations, but each accounts for only a few pedigrees worldwide. Others’ pathogenic significance remains unclear. Screening for LHON in a patient with visual loss should begin with the three primary mutations. In those primary mutation-negative patients in whom suspicion remains high, testing for the other mtDNA mutations associated with LHON is probably warranted, especially for those mutations deemed likely to be causal in a few pedigrees. Alternatively, since the majority of these other mtDNA mutations reside in genes encoding subunits of complex I, complete sequencing of complex I, perhaps beginning with the so-called “hot spot” ND6 gene, might also be considered. Finally, sequencing the entire mitochondrial genome is possible, although labor intensive. This should be performed only in those cases of high suspicion, and interpreted by someone versed in the complexities of mitochondrial genetics.
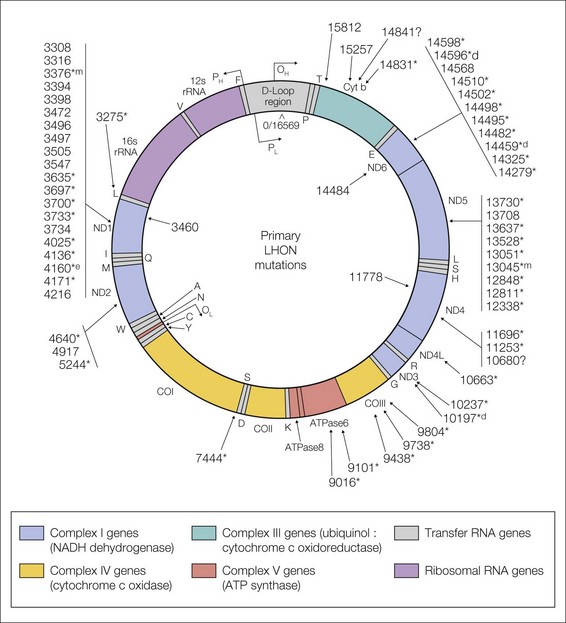
Among the primary mutations, the LHON clinical phenotype is remarkably similar. The only consistent differentiating feature is the better prognosis for visual outcome in those patients with the 14484 mutation.1 Up to 70% of patients with the 14484 mutation will have some degree of visual improvement compared to only 5% of patients with the 11778 mutation. Patients with the 3460 mutation may have a better chance of visual recovery than those with the 11778 mutation, but the numbers of patients are too small for meaningful analysis. Patients with an age of onset of visual loss of less than 20 years, especially less than 10 years, have a much better visual prognosis.2,3
The genetic defects, however, cannot fully explain the determinants of expression in this disease. The presence of a mtDNA mutation is necessary for phenotypic expression, but it is not sufficient.1–3 The presence of heteroplasmy (the co-existence of both mutant and normal mtDNA) may be a factor in expression. In heteroplasmic pedigrees, individuals with a greater amount of mutant mtDNA may be at higher risk for visual loss. However, many individuals with 100% mutant mtDNA never suffer visual loss. The interaction of genetic (mitochondrial or nuclear) and environmental factors may complicate the issue of assigning a pathogenic role to individual mtDNA mutations. Other mitochondrial or nuclear DNA factors may modify expression of the disease, including X-linkage as an explanation for the prominence of male expression. Various environmental triggers for the development of visual loss in LHON have been suggested. Systemic illnesses, nutritional deficiencies, and medications or toxins that stress the organism’s mitochondrial energy production might be detrimental to those individuals already genetically at risk for mitochondrial energy deficiency. Smoking, probably by its deleterious effect on mitochondrial function, has been associated with a higher expression of visual loss among carriers of the LHON primary mutation; only heavy alcohol intake has been similarly associated.5
The pathophysiology of LHON remains unknown.2–4 It may involve abnormal oxidative phosphorylation and deficient generation of ATP, either directly or indirectly related to free radical production, resulting in irreversible damage to the ganglion cells and their axons. The reason these mechanisms result in selective damage to the optic nerve remains uncertain. Histochemical studies of the optic nerve in animals have shown a high degree of mitochondrial respiratory activity within the unmyelinated, prelaminar portion of the optic nerve, suggesting a particularly high requirement for mitochondrial function in this region. Of great interest is the recent identification of genetically induced rodent models of complex I deficiency that show histopathological features of optic nerve degeneration similar to those seen in LHON patients.4 Further development of animal models for mitochondrial diseases should better facilitate the understanding of the pathogenesis of human mitochondrial disorders.
Therapies tried in the treatment of LHON include coenzyme Q10, idebenone, l-carnitine, succinate, dichloroacetate, vitamin K1, vitamin K3, vitamin C, thiamine, vitamin B2, and vitamin E.1–4 A randomized, controlled study of idebenone in the treatment of LHON patients within 5 years of visual loss suggested that patients with a disparity in their visual function between the two eyes (i.e. those likely earlier in the course of their disease) may have a better visual outcome with treatment.6 However, irreversible damage makes optic atrophy an unlikely candidate for a good response to any therapy. Avoiding agents that might stress mitochondrial energy production is a non-specific recommendation with no proven benefit. However, we suggest that LHON patients and maternal relatives at risk avoid tobacco, cyanide-containing products, excessive alcohol, and environmental toxins. Symptomatic therapies include pacemakers in those patients with heart block or serious cardiac conduction defects and low-vision aids for the patient with severe visual loss. As the specific genetic and biochemical abnormalities are better defined, more directed therapies may be created to replace or bypass the genetic or metabolic deficiencies in patients with the disease and in their relatives at risk. A promising form of gene therapy known as allotypic expression may play a future role in the therapy of LHON and other mitochondrial diseases.4 Meanwhile, one should not underestimate the importance of informed genetic counseling regarding maternal inheritance among family members.
Dominant optic atrophy
Autosomal dominant (“Kjer type”) optic atrophy (DOA) is the most common of the autosomal hereditary optic neuropathies, with an estimated disease prevalence in the northeast of England of 1 in 35 000 individuals, and as high as 1 in 12 000 in Denmark.1–3
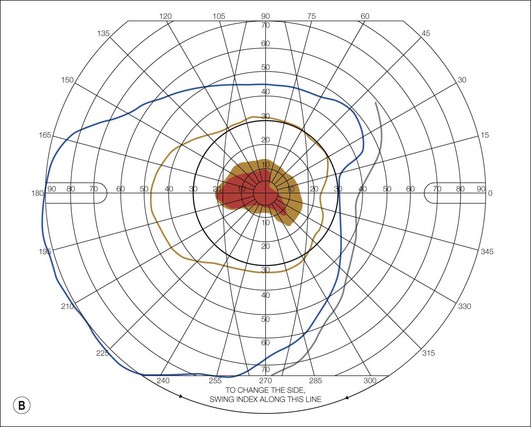
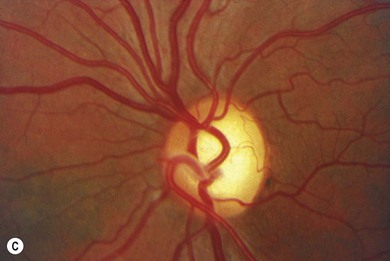
Although it is difficult for patients to identify a precise onset of reduced vision, the majority of affected patients date the onset of visual symptoms between 4 and 10 years of age, with 58−84% of patients reporting visual impairment by age 11. Rarely, severely affected individuals have visual difficulties and sensory nystagmus prior to schooling. Many patients are unaware of a visual problem and are discovered to have optic atrophy as a direct consequence of examination of other affected family members. These phenomena attest to the usually imperceptible onset in childhood, mild degree of visual dysfunction, absence of night-blindness, and absence of dramatic progression.1,2 Visual acuity is usually reduced to the same mild extent in both eyes. Acuities range from 20/20 to 20/800, with only about 15% of patients with vision of 20/200 or worse. Hand motion or light perception vision is extremely rare. There is considerable interfamilial and intrafamilial variation in acuities. Although not as rapid and devastating as LHON, DOA results in visual impairment severe enough to preclude safe motor vehicle operation in about 50% of affected adults. Blue-yellow color defects are classic in patients with dominant optic atrophy, but a mixed color deficit is most common. Visual fields in patients with dominant optic atrophy characteristically show central, paracentral, or cecocentral scotomas. A bitemporal depression may mimic chiasmal compression. The optic atrophy in patients with dominantly inherited optic neuropathy may be subtle, temporal only, or involve the entire disc (Fig. 52.3). The most characteristic change is a striking triangular excavation of the temporal portion of the disc, sometimes leading to the erroneous diagnosis of glaucoma. Insidious progressive decline in visual function occurs in about 50−75% of patients.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


