Hereditary Macular Dystrophies
Kenneth G. Noble
The hereditary macular dystrophies are a group of diseases that share certain similar features. In-herited in a monogenic fashion, all the typicalmendelian modes (autosomal dominant, autosomal recessive, and X-linked recessive) have been documented.
The most common presentation is a history of slowly progressive central vision loss occurring in the first 3 decades of life associated with a bilateral, often symmetric maculopathy. A careful family history should be taken, and appropriate family members should be examined. The retinal abnormalities are confined to the macula or posterior pole. Visual function tests will indicate the retina outside the macula to be normal.
DIFFERENTIAL DIAGNOSIS
Hereditary macular dystrophies must be distinguished from acquired macular degenerations because of the familial considerations and from generalized chorioretinal dystrophies because of the more dire prognosis.
Acquired macular degenerations usually occur later in life (beginning in the fifth decade) and are more likely to be unilateral or, if bilateral, asymmetric. Generalized chorioretinal dystrophies can mimic a macular dystrophy with regard to the history and fundus appearance, but tests of general retina function (e.g., the electroretinogram [ERG]) will indicate more widespread dysfunction. Generalized chorioretinal dystrophies that may mimic macular dystrophies include rod-cone dystrophy (retinitis pigmentosa), cone-rod dystrophy (inverse retinitis pigmentosa), progressive cone dystrophy, Leber’s congenital amaurosis, idiopathic juvenileX-linked retinoschisis, and Goldmann-Favre syndrome. These diseases are not discussed in this chapter.
CLASSIFICATION
Various methods have been used to classify the hereditary macular dystrophies, but none are entirely satisfactory. The limitations of categorization simply according to mode of inheritance or age at onset are evident. Currently, the preferred classification is based on the presumed anatomic location of the primary pathologic process. This is determined by the clinical examination and histopathologic correlation.
From a practical standpoint, it is important for the clinician to place these disorders in perspective based on the frequency of occurrence. The disorder of Stargardt’s disease and fundus flavimaculatus is easily the most common hereditary macular dystrophy and affects approximately 70% of patients with hereditary macular dystrophies whom I examine. The second most frequent dystrophy is Best’s vitelliform macular dystrophy. These two disorders account for nearly 90% of hereditary macular dystrophies. Dominant drusen of Bruch’s membrane, pattern dystrophies of the retinal pigment epithelium (RPE), and central areolar choroidal dystrophy are decidedly less common, and the remaining dystrophies are limited to a few reports in the literature.
STARGARDT’S DISEASE (JUVENILE MACULAR DEGENERATION) AND FUNDUS FLAVIMACULATUS
There is no consensus as to whether Stargardt’s disease and fundus flavimaculatus are two distinct diseases, perhaps with some overlap, or whether they represent different phenotypic manifestations of the same genetic disease. The typical appearance of Stargardt’s disease, described and depicted in Stargardt’s initial publications,1,2,3,4,5 is a pigmentary maculopathy surrounded by yellowish white spots. The typical appearance in fundus flavimaculatus6,7 is these yellowish spots or flecks occupying the entire posterior pole out to the midperiphery, with or without a pigmentary maculopathy. Studies of large groups and families8,9,10 and long-term evaluations suggest a continuum between the typical appearances of each disease.
Stargardt’s disease-fundus flavimaculatus, as the typical and most common macular dystrophy, serves as the prototype of this group in much the same fashion as retinitis pigmentosa is the paradigm for the generalized chorioretinal dystrophies. The common presentation occurs in a youngster who is referred because of failing a vision test or who complains of diminishing central visual acuity. At this time, the fundus examination shows a bilateral symmetric maculopathy. Tests of general retinal function (e.g., the ERG) are normal, and the course is gradual visual deterioration to levels of 20/200 to20/400. The mode of inheritance is autosomal recessive, and there is no sex, racial, or ethnic predilection.
Although the preceding description pertains to most patients, there are variations. The autosomal recessive mode of inheritance has been firmly established. There are, however, at least two well-documented dominant pedigrees that express the fundus manifestations of this disease.11,12 The age at onset of symptoms may actually occur at any decade of life depending on the severity of vision loss and perceptiveness of the patient. Visual acuity may be good and remain so despite the increasing disabling symptoms of paracentral central scotoma. I have seen six symptomatic persons in the fourth to sixth decades of life with 20/20 vision, severe and disabling paracentral scotoma, and a typical fundus appearance of this disease.
The fundus morphology at the time of presentation will be bilateral and symmetric, yet it can be varied. There may be a mild granularity of the macula without flecks, diffuse flecks without a maculopathy, circumscribed choroidal atrophy, a bull’s-eye appearance, bone spicule pigment clumping, or rarely, subretinal choroidal neovascularization. The flecks can vary in size, shape, and color; they are continually disappearing and reappearing elsewhere, sometimes leaving no trace, and at other times resulting in pigmentary or choroidal atrophy (Fig. 1). The ERG is the single most important test to differentiate a general retinal dystrophy from a local macular dystrophy. The ERG is usually normal in a local disease, but a decrease in the photopic b-wave response with a normal implicit time can be seen.9,10 A more advanced stage has been described in older persons in whom vision is worse than 20/400, there is evidence of diffuse chorioretinal abnormalities, and the cone and rod ERG responses are moderately or profoundly abnormal.10,13
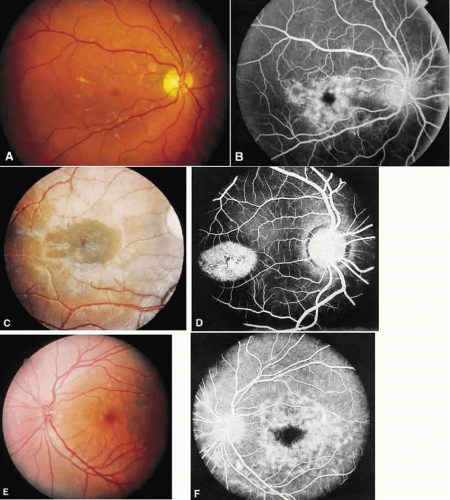 Fig. 1. Stargardt’s disease-fundus flavimaculatus. A. The typical characteristic fundus appearance is a mild macular pigment granularity surrounded by yellowish white flecks. B. On the fluorescein angiogram, these flecks show hyperfluorescence or hypofluorescence, and the background choroidal fluorescence is normal. C and D. A 13-year-old boy had a mild pigment granularity without flecks in both eyes associated with 20/200 vision. Although the granularity appears mild, the fluorescein angiogram shows a large central area of transmitted hyperfluorescence. The silent, dark choroid is evident here. The background choroidal fluorescence is markedly diminished and permits visualization of the retinal capillaries. E and F. A 36-year-old man was asymptomatic with 20/20 vision in each eye. The entire posterior pole and area nasal to the disc show irregularly shaped yellowish white flecks. A fluorescein angiogram shows areas of transmitted hyperfluorescence, many of which do not correspond to the flecks. Despite the widespread flecks, the background choroidal fluorescence is normal. |
The three histopathologic reports of this disease agree there is an accumulation of an abnormal material in the RPE but disagree as to its nature. The earliest study mentioned hyaluronidase-sensitive acid mucopolysaccharide, which is positive for periodic acid-Schiff.14 The two more recent studies cite a lipofuscinlike material and tubulovesicular lipid membranes.16
In view of the histology of an abnormal material in the RPE, the observations that more than 80% of patients who have had fluorescein angiography will show a decrease in the underlying choroidal fluorescence and an increase in the visibility of the retinal capillaries (a silent18 or dark19 choroid) warrants special attention. It has been postulated that the abnormal accumulation of lipofuscinlike material in the RPE may be responsible for the silent dark choroid because this pigment will absorb the underlying transmitted choroidal fluorescence.15 I have seen two brothers who initially showed a normal choroidal fluorescence. Eight years later, the eyes of one brother remained unchanged, but the other brother developed a silent dark choroid. The change may be the result of increasing amounts of lipofuscin accumulation.
The differential diagnosis depends on the fundus appearance. With regard to the maculopathy, an atypical morphology suggests diseases such as central areolar choroidal dystrophy, progressive cone dystrophy, vitelliform macular dystrophy, the later atrophic stages in X-linked retinoschisis, and various acquired macular degenerations. The yellowish white flecks may be confused with drusen of Bruch’s membrane, fundus albipunctatus, retinitis punctata albescens, multiple vitelliform cysts, and pattern dystrophies of the RPE. The mode of inheritance and visual function tests is helpful in distinguishing among these various disorders.
BEST’S VITELLIFORM MACULAR DYSTROPHY
Best’s vitelliform macular dystrophy is the second most common hereditary macular dystrophy but, for many reasons, is the most atypical and enigmatic. It has a variable age at onset, having been noted shortly after birth20 or newly arising in the sixth decade of life.21 Vision varies widely and may actually improve considerably with age. The fundus morphology has many appearances. It may be markedly asymmetric or even uniocular22,23,24,25,26,27,28,29, and in some affected patients with the mildest manifestations, may appear completely normal.30
The solid yellow egg yolk macular lesion is one of the most striking in all of ophthalmology. Unfortunately, this finding is not common in vitelliform macular dystrophy nor is it pathognomonic for this disorder. When it does occur, the patient is usually asymptomatic with 20/20 vision. When this egg yolk “ruptures,” the vision will then diminish. The appearance of the “scrambled egg” or “pseudohypopyon” maculopathy are now well recognized. However, the presenting fundus morphology of drusen, subretinal hemorrhage, choroidal neovascularization, choroidal atrophy, subretinal and intraretinal fibrosis, and nonspecific pigment atrophy and clumping will not suggest the correct diagnosis. When one also considers that the lesions may be multifocal, extramacular, asymmetric, and uniocular, establishing the diagnosis of vitelliform macular dystrophy can be most difficult (Fig. 2). In this regard, it is important to recall that in this autosomal dominant disorder, one of the parents is affected. Actually, all family members should be examined to establish the mode of inheritance since asymptomatic affected persons may show macular and extramacular manifestations.
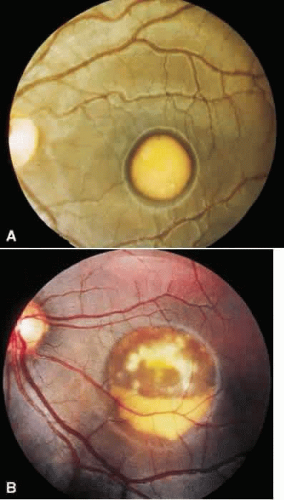 Fig. 2. Best’s vitelliform macular dystrophy. A. The solid egg yolk lesion is the classic appearance and is associated with normal vision. B. When the yellowish material begins to resorb, vision will decrease, as occurred in this 40-year-old woman. The outline of the cyst can be seen. Above is the appearance of the “scrambled egg,” and below is the layering of material in a “pseudohypopyon” appearance. |
The electrooculogram (EOG) is the one crucial diagnostic test for this disorder because there is a dichotomy between a normal ERG and an abnormal EOG.31,32 Studies of a large pedigree33 and many affected patients34 have indicated that the EOG is invariably abnormal in all affected members; the EOG is abnormal in both eyes of uniocular cases; and the EOG is abnormal in affected patients with a normal fundus. An abnormal EOG is therefore a sensitive indicator for the dystrophy and may be the only evidence of the inheritance of the abnormal gene.
The three most recent histopathologic studies all found an abnormal accumulation of lipofuscin within the RPE throughout the fundus.35,36,37 These studies, however, disagree as to the primary site of pathology, that is, the RPE36,37 or the sensory retina.35
Pseudovitelliform lesions are a heterogeneous group of disorders in which a yellowish macular lesion resembles the appearance seen in the hereditary macular dystrophy. Unlike true vitelliform dystrophy, however, these patients are older, have no evidence of a dominantly inherited disorder, and, most importantly, always have a normal EOG.33,34,35,36,37,38,39,40,41 These mimicking lesions have been associated with perifoveal retinal capillary leakage,39 RPE detachments,40 and nonspecific pigment changes.40,41 Therefore, these yellowish lesions probably represent an unusual morphology of an acquired macular degeneration.
DOMINANT DRUSEN OF BRUCH’S MEMBRANE
Dominant drusen of Bruch’s membrane is usually asymptomatic, and therefore, its true frequency is certainly higher than the frequency with which it is diagnosed. The relationship between drusen and age-related macular degeneration is well established clinically42,43 and histopathologically.43,44,45 However, on clinical and histopathologic grounds, there is reason to believe that these degenerative drusen are different from the dominantly inherited dystrophic drusen.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


