Essentials of Diagnosis
General Considerations
Hearing loss is the most common sensory deficit in humans. The prevalence of congenital hearing loss in newborns is approximately 1–3 cases per 1000. More than 60% of these prelingual cases (ie, hearing loss before the acquisition of speech) are attributed to genetic causes. A further 1 in 1000 children becomes deaf before adulthood. In patients over 60 years of age, approximately half show a hearing loss >25 dB HL. A large percentage of these populations is estimated to be likely affected by genetic influences, although age-related epidemiologic studies of the genetic contribution to hearing loss are not available. Finally, more than 100 deafness genes are believed to exist. These figures illustrate the impact of hearing loss on the public health system and the importance of genetic factors.
Classification
The most common and useful distinction in hereditary hearing impairment is syndromic versus nonsyndromic hearing impairment. Seventy percent of hereditary hearing impairments are nonsyndromic, whereas a minority of 15–30% are syndromic (Figure 54–1).
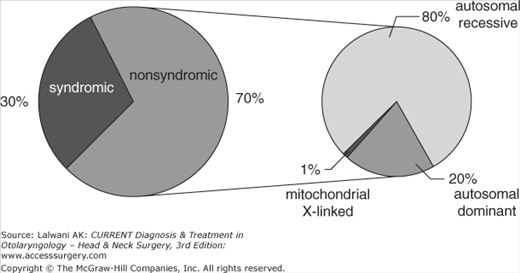
Nonsyndromic hereditary hearing impairment is classified by the mode of inheritance. Autosomal recessive transmission (designated by prefix DFNB) is implicated in approximately 80% of cases, autosomal dominant transmission (DFNA) is present in approximately 20% of cases, and X-linked (DFN) and mitochondrial transmission are responsible for <2% of cases (see Figure 54–1). One single gene, GJB2 (Gap-Junction Beta 2 encoding for connexin 26), has emerged to be the most common cause of recessive deafness, and up to 40% of the onset of sporadic prelingual hearing impairment can be attributed to defects in this gene both in Europe and the United States. The prevalence is higher in southern Europe than in northern Europe, mainly owing to one single gene mutation, c.35delG. In a stretch of six guanines extending from position 30 to 35, one base pair is deleted. The high incidence of this mutation seems to be due to a common ancestor. Other common mutations include c.167delT in Ashkenazi Jews and c.235delC in the Japanese population. Also, a common digenic pattern of inheritance involving GJB2 and GJB6 has been detected. Patients with a monoallelic mutation in GJB2 harbor in addition a deletion of GJB6.
Mitochondrial genes constitute a small and unique group. Inheritance is entirely through the mother, because the maternal oocyte is the sole contributor of mitochondria. Although hearing loss occurs frequently in mitochondrial diseases, it is much more seldom the only symptom. The c.A1555G mutation in the MT-RNR1 gene is the most important one among inherited hearing impairment with mitochondrial transmission.
Syndromic hearing impairment means that hearing loss is accompanied by other clinical abnormalities. More than 400 syndromes that include hearing loss have been described in detail. Currently, syndromic hearing loss is categorized as follows: (1) syndromes due to cytogenetic or chromosomal anomalies; (2) syndromes transmitted in a classic monogenic or mendelian inheritance; or (3) syndromes due to multifactorial influences, in which the phenotype results from a combination of genetic and environmental factors. The breakdown of the genetic code of the various syndromes will most certainly lead to a classification based on molecular-genetic findings.
Pathogenesis
Several distinctions are typical of hereditary hearing impairment. Usually, the disease is genetically highly heterogeneous, with many different genes responsible for auditory dysfunction. To complicate things further, different mutations in one gene can cause variable phenotypes (eg, connexin genes in the skin or the ears) or even syndromic and nonsyndromic hearing loss as seen in genes SLC26A4 (Pendred syndrome and DFNB4), MYH9 (May-Hegglin/Fechter syndrome and DFNA17), and WFS1 (Wolfram syndrome and DFNA6/DFNA14). Finally, a mutated gene can cause dominant and recessive forms of hearing loss. Typical examples include GJB2 and TECTA.
Most of the nonsyndromic genes that cause deafness are not restricted to the cochlea; the inner ear seems to be more sensitive to disruption of some cellular functions than are other organs. In most cases, the function of these genes is only slightly understood. Several genes involved in ion homeostasis and cytoskeleton (ie, hair-cell) structure that lead to deafness have been identified. Other genes include cell-to-cell interaction, transcription factors, extracellular matrix and a few genes with unknown functions. Nonsyndromic genes discovered by the end of 2009 are listed in Table 54–1, which includes their function and mode of inheritance.
| Gene | Function | Transmission |
|---|---|---|
| KCNQ4 | Autosomal dominant | |
| WFS1 | ||
| CRYM | ||
| CLDN14 | ||
| TRIC | Homeostasis | Autosomal recessive |
| SLC26A4 | ||
| GJB2 (Connexin 26) | ||
| GJB3 (Connexin 31) | both | |
| GJB 6 (Connexin 30) | ||
| TMC1 | ||
| OTOF | Exocytosis of neurotransmitter | Autosomal recessive |
| MYH9 | ||
| ACTG1 | ||
| DIAPH1 | ||
| CCDC50 | Autosomal dominant | |
| USH1C | ||
| PCDH15 | ||
| MYO15 | ||
| TRIOBP | Cytoskeletal system | |
| MYO3A | ||
| THMS | ||
| SLC26A5 | ||
| WHRN | Autosomal recessive | |
| CDH23 | ||
| RDX | ||
| MYO7A | ||
| MYO6 | both | |
| ESPN | ||
| EYA4 | Autosomal dominant | |
| TFCP2L3 | ||
| POU4F3 | Transcription factors | |
| POU3F4 | X-linked | |
| ESRRB | Autosomal recessive | |
| COCH | Autosomal dominant | |
| OTOA | Autosomal recessive | |
| STRC | Extracellular matrix | |
| TECTA | both | |
| COLL11A2 | ||
| MYO1A | ||
| DFNA5 | Autosomal dominant | |
| MYH14 | Unknown | |
| TMPRSS3 | Autosomal recessive | |
| TMIE | ||
| PJVK |
In the homeostasis group, the gap-junction proteins (connexins) are the most well known. Three types of connexin genes have been discovered; GJB2 is the most prevalent. The protein encoded by GJB2 (connexin 26) is involved in intercellular transport of ions, metabolites, and second messengers. Based on its expression in the human cochlea in the stria vascularis, the basement membrane, the limbus, and the spiral prominence, as well as in animal studies, the role of GJB2 seems to lie in recycling potassium ions back to the endolymph of the cochlear duct after stimulation of the sensory hair cells.
OTOF is responsible for synaptic exocytosis of neurotransmitters in auditory hair cells.
Unconventional and conventional myosins represent the largest group of genes involved in hair-cell structure and motility. Myosins are actin-dependent molecular motors. Unconventional myosins are found in different locations in the inner ear, including the hair cells. Their various functions include endocytosis, the regulation of ion channels, the movement of vesicles in the cytoplasm, and anchoring stereocilia.
Transcription factors are important for regulating the expression of other genes. The EYA4 protein, for example, regulates the early development of the organ of Corti and maintains its continued function postdevelopmentally.
The list of genes responsible for syndromic hearing impairment encompasses diverse molecules, such as enzymes, transcription factors, and cytoskeletal and extracellular matrix components. Although syndromic deafness is mostly inherited in an autosomal dominant fashion, in some cases, the transmission from parents to children does not occur. A classic example is neurofibromatosis, in which approximately 50% of genetic mutations are spontaneous. A summary of genetic findings is listed in Table 54–2.
| Gene | ||||
|---|---|---|---|---|
| Syndrome | Clinical Findings | Name | Function | Localization |
| Pendred syndrome | Goiter | PDS | Anion exchanger | Endolymphatic duct and sac, utricle, saccule, cochlea |



