Chapter 57 Hemoglobinopathies
In a landmark 1910 Archives of Internal Medicine article,1 Dr James Herrick noted “peculiar, elongated, sickle-shaped red blood corpuscles” from a peripheral blood smear of a Grenadian dental student with recurring medical illnesses and, thus, penned the first written description of sickle-cell disease (SCD).1–3 Linus Pauling and colleagues in 1949 were the first to implicate a defective hemoglobin molecule within the erythrocyte as the cause of the disease.4,5
The term “sickle-cell disease” encompasses the group of hemoglobinopathies characterized by intravascular hemolysis and by defective oxygen transport. In a normal red blood cell, two α-globin subunits, two β-globin subunits, and a central heme molecule combine to form adult hemoglobin (Hb A). The β-globin gene, an oxygen transport gene, is found on the short arm of chromosome 11.6,7 Hemoglobin S (Hb S) results from a single amino acid point mutation in which a valine substitutes for a glutamic acid at the sixth position within the β-globin chain. Hemoglobin C (Hb C) is caused by a glutamic acid to lysine mutation in the β-globin molecule.
SCD is transmitted through the autosomal recessive mode of inheritance. Two copies of Hb S may combine with one another (SS disease), or one copy of Hb S and another β-globin variant, such as Hb C, may combine (double heterozygous SC disease).7 Individuals with one copy of Hb A and one copy of Hb S are described as having the sickle-cell trait, the carrier state for SCD. β-thalassemia occurs when a reduced amount of β-globin is present. This condition is beta-plus (β+) thalassemia, whereas the absence of β-globin is called beta-zero (β0). Either of these conditions may combine with Hb S, leading to a compound heterozygous state.7
Prevalence
To date, SCD remains the most common inherited blood disorder, and affects nearly 80 000 people of African American and Hispanic descent.8 SCD occurs in 1/500 African American births and every 1/36 000 Hispanic-American births.8–10 SCD results in 5% of deaths in children under 5 years in Africa, more than 9% of such deaths in West Africa, and up to 16% of under-5 deaths in individual West African countries.11 Approximately 8% (or one in 12) of black Americans possess sickle-cell trait, which is not associated with increased mortality or morbidity (except in hyphema), and is thought to confer a genetic protection against malarial infection.10 Their hemoglobin concentration is typically 35–40% Hb S and 55–60% Hb A.7 Although sickle-cell trait subjects are generally asymptomatic, they may encounter systemic complications of SCD under conditions of extreme hypoxia.12 Populations of African descent possess the highest frequency of Hb S. At-risk genotypes for SCD are also observed in people of Mediterranean, Caribbean, South and Central American, Arab, and East Indian descent.7 SS disease is displayed in approximately 0.15% of African descendants in North America.7,13 Those with SS disease do not typically become symptomatic until after 6 months of age, when the fetal hemoglobin (Hb F) is replaced with Hb S. These individuals have a decreased life expectancy, as they are prone to developing severe anemia and are highly susceptible to recurrent infections.14 Individuals with Hb SC disease, the double heterozygotes, typically exhibit 50% Hb S and 50% Hb C hemoglobin composition and typically display less morbidity from the systemic complications of SCD, and have a normal life expectancy.7 With a hemoglobin composition of 60–90% Hb S and 10–30% Hb F, sickle β-thalassemia patients are typically encountered in central African and Mediterranean countries. Those with other forms of sickle β-thalassemia may have Hb A as 10–30% of their hemoglobin composition.7,15
Genetic modifiers
The degree of phenotypic expression of SCD is quite variable, even among those with the same genotype. Environmental effects and multiple gene interactions are thought to be at play. Further, the ability of an individual to generate Hb F may lead to reduced disease severity. Different β-globin cluster haplotypes may result in different levels of Hb F.7,16 Levels of Hb F have been shown to correlate with the clinical manifestations of SCD.17
Pathophysiology
The interplay among abnormal erythrocytes and hypoxic, hyperosmolar, or acidotic conditions leads to the abnormal rheology and hemolysis characteristic of SCD. In Hb S, a strongly hydrophobic polar valine takes the place of a nonpolar strongly hydrophilic glutamic acid residue.5 Upon deoxygenation in the microcirculation, hydrophobic residues within Hb S are exposed and associate with hydrophobic regions of adjacent molecules. This polymerization results in the generation of rigid fibers of Hb S which damage the red blood cell membrane and cytoskeleton and cause the cell to assume a sickle shape. This polymerization process is reversible as oxygenation increases, and the cell may resume its native discoid shape. However, the repeated cycle of sickling and unsickling of the red blood cell membrane may lead to permanent damage to the erythrocyte membrane, irreversible sickling, and hemolysis. Mean corpuscular hemoglobin concentration (MCHC) may be the most important factor contributing to the rate of Hb S polymerization.5 The higher the MCHC, the more hemoglobin molecules that are available to participate in polymerization, and the closer these molecules are to one another, further promoting a favorable environment for Hb S polymerization.18,19
The erythrocyte’s original state of oxygenation also impacts the extent and rate of polymerization.15,16 An intrinsic property of the normal erythrocyte is the ability to deform easily in order to pass through capillaries with smaller diameters than its own. Decreased erythrocyte deformability and increased rigidity can cause increased capillary transit time.19 Deoxygenation and sickling promote increased permeability of the cell membrane to potassium, sodium, and calcium cations, which leads to water efflux from the cell, cellular volume contraction, and resultant increase in Hb S concentration.5,19,20,21
In addition to mechanical obstruction of blood vessels by dense, sickled erythrocytes, these sickled erythrocytes display increased adhesion to vascular endothelium matrix proteins, such as laminin,22,23 and thus cause direct damage to the endothelium. Integrin α4β1, integrin-associated protein, sulfated glycolipid, lutheran protein, phosphatidylserine, band 3 protein, and CD36 are adhesion molecules expressed in sickled red blood cells.24–26 Immature red blood cells called reticulocytes increase in SCD following intravascular hemolysis. These cells also have increased adhesion molecules, such as integrin α4β1,27–29 which promote pathologic adhesion to the vascular endothelium, specifically to vascular cell adhesion molecule-1 (VCAM-1). Direct activation of endothelial cells occurs in response to elevated circulating cytokines such as tumor necrosis factor-α (TNF-α) and interleukin-1β (IL-1β),30,31 which upregulate expression of endothelial adhesion molecules like intercellular adhesion molecule-1 (ICAM-1), VCAM-1, E-selectin, and P-selectin.32,33
Inflammation likely plays a role in the vaso-occlusive process in SCD. Lutty and colleagues demonstrated retention of SS red cells and adherence of red cells in reticulocyte-rich fractions in retina and choroid of rat eyes in hypoxic conditions or following TNF-α stimulation.34–36 TNF-α and IL-1 may contribute to vaso-occlusion by accelerating the production of adhesion molecules on the vascular endothelium and by activating polymorphonuclear leukocytes.30 TNF-α and IL-1 have been shown to be upregulated in the sera of individuals with SCD at baseline.31,37,38
Nitric oxide (NO) is a potent vasodilator and regulator of vascular tone; it is derived from the vascular endothelial NO synthase. SCD has been associated with elevated reactive oxygen species, which scavenge NO and metabolize arginine, its precursor.7 l-arginine as an oral supplement has been given to induce NO production in transgenic sickle-cell mice.38,39
Vascular endothelial growth factor (VEGF), which is upregulated by hypoxia, is present in the serum of SCD patients at baseline, and is elevated during vaso-occlusive crises.40 Elevated VEGF has been demonstrated in eyes with sickle-cell retinopathy.41,42 VEGF has also been shown to increase levels of cell adhesion molecules, ICAM-1 and VCAM-1.43,44 Angiopoietin-1 (Ang-1) and angiopoietin-2 (Ang-2) interact with the Tie-2 receptor on endothelial cells, regulating angiogenesis. Ang-1 is responsible for the maintenance and stabilization of mature blood vessels, while Ang-2 leads to vessel destabilization and dissociation of pericytes and is upregulated by hypoxia and VEGF.45 Interaction among these proteins may be important in the pathogenesis of SCD.
Systemic manifestations
Over recent years, as the life expectancy of those affected by SCD has increased, so have the number of complications these individuals experience.12,46 Sickling of the red blood cells, with resultant intravascular hemolysis, thrombosis, tissue necrosis, and ischemia, causes a myriad of systemic complications, including cerebrovascular accident, acute chest syndrome, pulmonary hypertension, splenic sequestration, priapism, osteonecrosis, cholelithiasis, pneumonia, leg ulcers, aplastic crisis, renal disease, need for recurrent transfusions, episodic, painful vaso-occlusive crises, and death.7
Severe visual impairment and blindness from complications of proliferative sickle-cell retinopathy (PSR) may also occur. For reasons that are as yet unclear, subjects with Hb SC disease typically display less systemic morbidity from their hemoglobinopathy, but have a higher likelihood of experiencing the ophthalmic complications, than their homozygous Hb SS counterparts.47 In one study of patients with SCD, retinopathy was detected in 33% of patients with Hb SC disease compared to 3% of patients with Hb SS disease.37
Several theories have been proposed to explain this difference. One explanation could be that Hb SS patients typically have a shorter life expectancy than Hb SC patients, and may not live long enough to manifest the ophthalmic disease. Alternatively, the difference in retinopathy among sickle-cell genotypes may be due to the higher hematocrit and cell density, and the lower Hb F, of individuals with Hb SC as compared to Hb SS individuals.7 The overall lower hematocrit in Hb SS patients, and the resultant lowered viscosity of blood, may confer a relative protection from vaso-occlusion in the retinal circulation.48 Another theory is that the retinal vaso-occlusions in SS disease may be so complete that no viable tissue remains to produce the growth factors necessary to mount a proliferative response. In Hb SC disease, the vascular occlusions may be less complete, and they occur in the setting of a chronic hypoxic rather than anoxic state. Thus, a steady-state release of angiogenic growth factors from the remaining viable tissue may occur.49 Finally, Lutty and colleagues showed that irreversibly sickled Hb SS erythrocytes are easily trapped in retinal capillaries and precapillary arterioles in hypoxic conditions in a rat model.16,35 Hb SC cells, however, regardless of oxygen levels, were less easily trapped in the retinal microvasculature. When the vascular endothelium was stimulated with cytokines, retention of Hb SC cells did occur. The authors suggested that vaso-occlusion in Hb SC disease may be the result of the complex interaction among cytokines, the fibrinolytic cascade, leukocyte interactions, and activation of the vascular endothelium with induction of adhesion molecules.13,35 This data suggests that retention of sickled cells in the retina is not mechanical (rigid cells retained in a small lumen) but rather due to hypoxia-initiated cytokines and receptors.
Ophthalmic clinical features
Retrobulbar and orbital involvement
Orbital involvement of SCD is uncommon, but has been reported. One study from Oman reported periorbital swelling during vaso-occlusive crises in five patients.50 The patients ranged in age from 6 to 15 years old. Four of these patients had SS disease, while one had sickle-beta thalassemia. Infarction of orbital bones and orbital hematomas occur, and may lead to the orbital compression syndrome. These patients typically display lid edema, fever, facial pain, proptosis, restriction of motility, and resultant diplopia. The authors concluded that these syndromes usually resolve with conservative management with treatment of the underlying crisis, antibiotics, and steroids. Orbital involvement in SCD may cause sudden permanent unilateral loss of vision without detectable retinal arterial changes due to retrobulbar ischemic optic neuropathy.7,51 Urgent decompression of the orbit may be required. Recurrent bilateral lacrimal gland swelling has also been reported.52
Anterior-segment involvement
Conjunctival and iris findings may be present in SCD. An early reported finding is the saccular and sausage-like dilatations of the tiniest conjunctival vessels.53 They are most easily seen with the highest power of the slit-lamp biomicroscope. Paton described these comma-shaped capillary segments in the inferior bulbar conjunctival vessels, and coined the term “conjunctival sign.”54 This sign was uncommon in those with high Hb F levels. Local heat from the slit lamp induces vasodilation, and causes reversal of the comma-shaped vessels,55,56 whereas topical vasoconstricting drops increase the abnormality.57,58 These conjunctival vascular changes vary with oxygenation status, and are more prevalent in Hb SS disease than in Hb SC disease.54 Pathophysiology studies show constriction of these conjunctival vessels during painful crises, with normal blood flow returning on recovery.58 On histopathologic examination, these vessels exhibit endothelial proliferation, dilatation, and thinning of the proximal blood vessel segments, and aggregation of red blood cells in the distal portions of capillaries.58
Segmental iris atrophy and pupil abnormalities can be seen in individuals with SCD (Fig. 57.1).59,60 Iris atrophy is thought to result from sectoral ischemic necrosis involving radial iris vessels. Secondary neovascularization of the iris stroma has also been described and documented with intravenous fluorescein angiography, and in one instance this neovascular frond resembled that classically seen as the “sea fan” in PSR.13,61 Neovascular glaucoma rarely occurs as a consequence of PSR.7,62
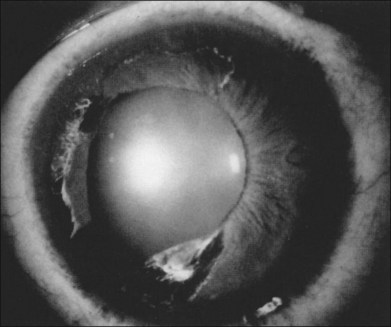
Hyphema in a patient with SCD and in those with sickle-cell trait represents a sight-threatening emergency, as even modest elevations of intraocular pressure (IOP) have resulted in vision loss from central retinal artery occlusion or macular branch retinal artery occlusion.62–65 This is likely a mechanical phenomenon, as sickled red blood cells clog the trabecular meshwork,61 causing elevation in IOP. Although intensive medical management to lower IOP in SCD patients with hyphema is recommended, IOP-lowering medications must be used cautiously. Repetitive use of carbonic anhydrase inhibitors, for example, is contraindicated in SCD patients with hyphema as most of these medications cause intracameral acidotic conditions that may worsen erythrocyte sickling. Early surgical intervention for IOP control should be considered, typically by paracentesis of the anterior chamber.66 Intracameral tissue plasminogen activator may be a potentially useful agent in the treatment of posttraumatic hyphema in some cases.67 Hyperoxygenation of the patient may also be helpful.
Posterior-segment involvement
Vitreoretinal interface
The pathologic retinal neovascularization caused by retinal ischemia in SCD can result in vision loss from vitreous hemorrhage.68,69 Other abnormalities of the vitreoretinal interface have been described, such as peripheral retinal whitening, which may represent abnormal or particularly strong adhesion at the vitreoretinal interface. This has been described in 93% of Hb SS patients,70 83% of Hb SC patients,71 and 82% of Hb S Thal patients.72 There is no known pathologic significance to this finding. This peripheral whitening is noted in the peripheral retina in the normal population as “white without pressure,” and may not be truly unique to those with sickle-cell retinopathy.13,57
Correspondingly, flat, brown, ovoid lesions in the retinal periphery have been noted in SCD, and termed “dark without pressure.” These lesions can be transient, and may disappear without sequelae. The underlying choroid appears normal on ophthalmoscopy, and the corresponding fluorescein angiogram is also normal.13,73
Optic nerve
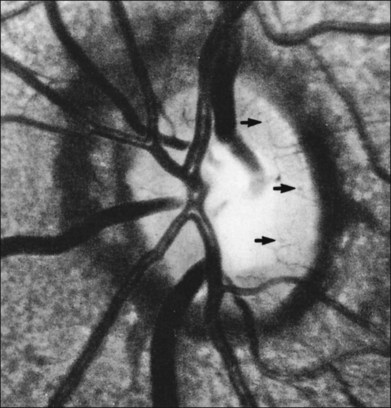
The optic nerve in patients with SCD may exhibit vascular changes. Dark, dilated capillaries at the optic nerve head appear as small red dots (Fig. 57.2), and represent precapillary arterioles plugged with sickled erythrocytes. These abnormal vessels have a linear or Y-shaped configuration on high-magnification ophthalmoscopy.13,57,70,74 These changes are not visually significant as the vascular occlusions are transient. Optic disc neovascularization is rare but has been reported in four Hb SC patients13,75–78 and one Hb SS patient.13,70
Macula
Though the hallmark of ocular SCD has been the peripheral retinopathy, the macula is also susceptible to infarction from vaso-occlusive disease. The “macular depression sign” has been described as an oval depression of the bright foveal or parafoveal reflex as a result of macular thinning due to ischemic atrophy.79 This finding is best detected using red-free illumination. The extent of macular vascular changes has not been shown to correlate with visual acuity. In a study by Sanders and colleagues, an enlarged foveal avascular zone (FAZ) was identified in patients with SCD on fluorescein angiography as compared to healthy, age-matched controls.80 Within the sickle-cell group, visual acuity did not correlate with FAZ size. The authors reported an enlarged FAZ in patients with SCD irrespective of the type of hemoglobinopathy or degree of retinopathy.
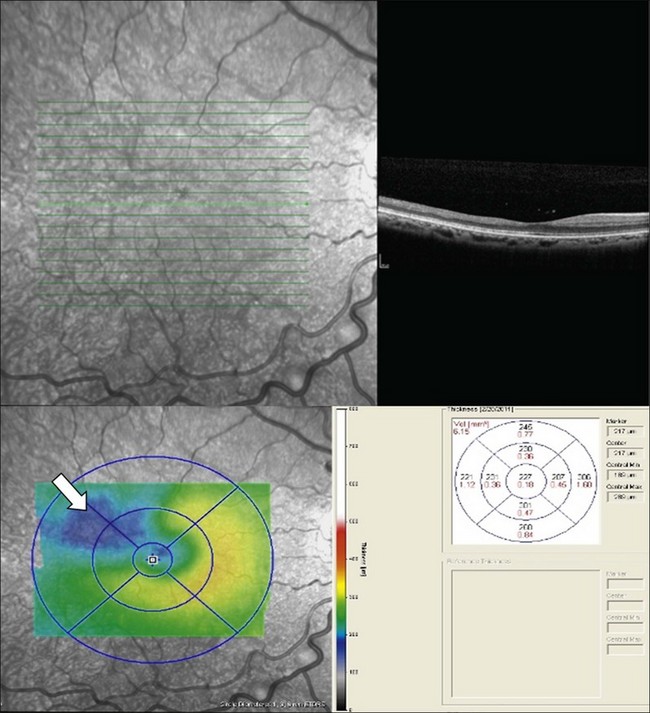
Macular infarcts can now be confirmed using optical coherence tomography.81 In a recent study utilizing spectral-domain optical coherence tomography (SD-OCT), Hoang and coworkers82 described retinal thinning of the central macula compared to healthy controls, most notably in the outer retinal layers in the central macula and parafoveal retina. Although several previous studies have reported atrophy of inner retinal layers after macular infarction, Hoang et al. hypothesize that the outer retinal layers may be thinned due to ischemia and resultant atrophy of the choriocapillaris, as these larger-caliber vessels may possibly be more prone to occlusion than the retinal vessels. Additionally, this study reported “splaying,” or blunting of the foveal contour on SD-OCT in asymptomatic patients with SCD with areas of focal parafoveal thinning (Fig. 57.3). A case series by Murthy et al. also describes thinning of the temporal macula in patients with PSR on SD-OCT, and suggests that this finding should lead clinicians to perform peripheral wide-field angiography to evaluate the retinal periphery for ischemia.83 Macular hole,84 epiretinal membranes,85 macular schisis,86 and posterior pole neovascularization87 have all been reported as rare complications of sickle-cell retinopathy.
Angioid streaks
Angioid streaks, breaks in Bruch’s membrane, have a well-documented association with SCD, occurring in 1–2% of patients.13,70,88 These irregular, reddish subretinal bands are most commonly found in patients with the Hb SS genotype.70,89 The incidence of angioid streaks in these eyes increases with age, and one Jamaican study found that 22% of 60 patients with Hb SS disease over age 40 had angioid streaks, compared to 2% of 150 patients under 40.90 Why angioid streaks occur in SCD is unclear. One hypothesis is that elastic tissue injury may occur as the result of oxidative damage due to hypoxia, and inflammatory cells may release cytokines that cause tissue damage.7,91 Angioid streaks in SCD usually have a benign course and resultant choroidal neovascularization (CNV) is rare.13
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


