Head and Neck Tumor Biology
Alec Vaezi
Jennifer R. Grandis
The treatment of head and neck squamous cell carcinoma (HNSCC) evolved slowly over most of the 20th century, with important breakthroughs including the description of the radical neck dissection by George Crile (1) in 1906 with an improvement of 3-year survival from 19% to 75%, the report of modified radical neck dissection by Suarez et al. (2) in 1963, the introduction of postoperative adjuvant external beam radiotherapy in the 1970s, and the use of adjuvant chemotherapy in the late 1980s. However, the last 20 years have brought considerable advances in the understanding of head and neck cancer tumor biology. Molecular techniques helped epidemiologists identify and characterize human papillomavirus (HPV)-associated HNSCC, which is currently responsible for an epidemic of oropharyngeal HNSCC extending throughout North America and, to a lesser extent, Europe. In addition, with better understanding of the genetic and molecular changes that characterize HNSCC, novel therapeutic agents have been designed and used with success in the treatment of HNSCC, such as the EGFR-blocking antibody cetuximab (Erbitux). Furthermore, novel prognostic and predictive biomarkers are being evaluated in the clinical setting to inform personalized cancer treatments in which the treatment is tailored to the biology of an individual tumor. HNSCC vaccine trials are currently accruing patients, and the role of cancer stem cells (CSCs), angiogenesis, and microRNAs is under study. Very recently, the HNSCC cancer genome has been outlined by sequencing the genomes or exomes of nearly 75 tumors (3). In sum, these times are exciting not only for head and neck tumor biologists but also for clinicians and patients who are already seeing the benefit of scientific discoveries in the clinical setting.
As basic science increasingly merges with clinical medicine, it is important for the clinician to be familiar with the different molecular pathways contributing to HNSCC development. A working knowledge of this biology has become essential for understanding treatment options for HNSCC in the 21st century. This chapter reviews selected major molecular pathways contributing to HNSCC. These include the mitogen signaling cascade, the cell cycle and its regulation, the major DNA repair pathways, and protection against apoptosis. The concept of solid tumor stem cells and angiogenesis is also discussed. For each section, a current model for the pathway is described, highlighting how deregulation of this pathway participates in tumor development. Examples illustrate how each pathway is involved in HNSCC tumor biology, and its clinical relevance. However, it is important to remember that basic science is in constant evolution. What appears meaningful today may be obsolete tomorrow. Every effort has been made to provide models backed by strong evidence that are likely to withstand the test of time.
DIFFERENT SUBTYPES OF HNSCC IDENTIFIED BASED ON RISK FACTORS AND GENETICS
Head and neck cancer is an important cause of death and morbidity worldwide. It is the eighth most frequent type of cancer in the USA with nearly 50,000 new cases diagnosed every year accounting for 11,000 deaths (4). Similar to other cancer types, HNSCC is proportionally even more deadly worldwide, where 510,000 new cases of HNSCC will be diagnosed yearly and more than half of these patients will succumb to their disease (5). Each of these cancers is the result of a multistep accumulation of genetic mutations in a progenitor cancer cell (6, 7, 8). Subsequent proliferation of mutated progenitor cells results in clonal expansion. With further accumulation of mutations, the clonal outgrowth becomes unregulated and irreversible. The nascent tumor progressively acquires new phenotypes including the capacity for invasiveness and metastasis. It was previously thought that head and neck cancers resulted from 6 to 10 independent significant genetic mutations acquired
over a period of 20 years (9). However, recent data from head and neck cancer genome sequencing suggest that many more mutations accumulate, although most of these may be of little consequence (i.e., passenger mutations) (3). In HNSCC, on average, 1 to 15 base pairs are mutated for each megabase of exome (3), which suggests there may be at least 6,000 to 90,000 mutations for the 6.4 billion base pairs in the human diploid genome.
over a period of 20 years (9). However, recent data from head and neck cancer genome sequencing suggest that many more mutations accumulate, although most of these may be of little consequence (i.e., passenger mutations) (3). In HNSCC, on average, 1 to 15 base pairs are mutated for each megabase of exome (3), which suggests there may be at least 6,000 to 90,000 mutations for the 6.4 billion base pairs in the human diploid genome.
Over the past 15 years, it has been increasingly evident that all tumors labeled as HNSCC may not represent the same clinical entity (10). For instance, HNSCC tumors have different prognosis depending on the affected site. A tonsil cancer from a middle-age smoker-drinker behaves differently than a lateral tongue cancer from an elderly woman who never smoked or drank, or a tonsil cancer from a young nonsmoker. Epidemiologic and clinical studies suggest that even HNSCCs with a similar histologic appearance may represent different types of tumors based on molecular heterogeneity, pathogenesis, risk factors, and clinical outcomes.
Exposure to carcinogens present in tobacco or betel quid is a major factor for head and neck cancer that has been recognized for years. Tobacco derivates contain a potent carcinogen, which damage the DNA and mutate the genome of progenitor cancer cell; tobacco smoke contains more than 4,000 toxic compounds including 81 carcinogens (11), and smokeless tobacco contains mutagenic nitrosamines (11). Interestingly, in vitro, one of the most potent carcinogens found in cigarette smoke, named benzo[a]pyrene diol epoxide, preferentially mutates p53 at hot spots found in lung cancer (12). It is therefore perhaps not surprising that p53 is the most frequently mutated gene in HNSCC, since tobacco consumption and synergistically excessive alcohol intake are the two major risk factors for HNSCC in the USA; “heavy” tobacco consumption increases the cancer risk by 5.8 times, “heavy” drinking by 7.4 times, and combination of the 2 increases the risk by 38 times in male and 100 times in females (13). In eastern countries, areca nut chewing in betel quid has been identified as a risk factor for HNSCC. The areca nut does not contain a single organic chemical responsible for the cancer formation, but rather a combination of chemicals that affect distinct steps of carcinogenesis. Areca alkaloids have genotoxic effects and participate in tumor initiation, and arecaidine contributes to tumor promotion (14).
After HPV was identified in oropharyngeal cancers (15, 16, 17, 18, 19), it became evident that a subset of oropharyngeal HNSCC is not associated with smoking and drinking but rather with promiscuous oral sex behavior and to seropositivity of sexually transmitted HPV16 (18, 20). HPV-positive HNSCC found in nonsmokers has a better prognosis than traditional HNSCC found in smoker/drinkers (21, 22). That these tumors represent two groups of HNSCC with distinct tumor biology is also evident at a genetic level: considerably fewer mutations are found in HPV-positive tumors from nonsmoker patients (3). Interestingly, retrospective analyses of archived tumors suggest that oropharyngeal HPV-positive tumors represent a relatively recent entity; also, to date, this epidemic has spread more extensively in the USA than in Europe. HPV-positive HNSCC in nonsmokers is now widely recognized as a distinct group of HNSCC (21). With further progress in the genetic and molecular understanding of HNSCC tumor biology, novel groups of HNSCC are likely to be characterized.
Other subtypes of HNSCC may originate on particular genetic backgrounds. Humans are not identical due to a certain genetic variability, or polymorphism, observed within the human population. In certain instances, the polymorphism has a profound effect on the function of a cancer susceptibility gene and induces a cancer-prone syndrome, sometimes an HNSCC-prone syndrome. For instance, patients with a congenital deficiency in one of the multiple genes coding for the Fanconi anemia DNA repair pathway proteins are 500 to 700 times more prone to HNSCC compared to other patients. Fanconi anemia patients, recognized at birth by finger aplasia, abnormal facies, and short stature, develop childhood aplastic anemia due to apoptosis of hematopoietic cells, and myelodysplastic syndrome in adolescence, with an 800-fold increased chance of myeloid leukemia (23). These patients are exquisitely sensitive to genotoxic stress by cross-linking agents (such as mitomycin C) because they cannot efficiently repair mutagenic DNA interstrand cross-links; as a consequence, their cells rapidly accumulate mutations when exposed to carcinogens (23). In a similar vein, it has been proposed that polymorphisms in DNA repair genes that mildly affect DNA repair capacity may lead to increased susceptibility to HNSCC due to hypersensitivity to environmental DNA-damaging agents. However, increased HNSCC susceptibility due to polymorphisms in DNA repair genes remains largely unproven (24).
PATHOGENESIS AND FIELD CANCERIZATION
Most of our knowledge about HNSCC pathogenesis comes from oral cancer. Precursor lesions of oral SCC are oral leukoplakia and erythroplakia. Clinically evident premalignant lesions are prevalent in less than 0.5% of the population where the rate of progression to cancer is estimated to be 1% to 2% per year (25). The leukoplakia is an exclusion diagnosis. It is a clinical term referring to a patch of white mucosa that does not disappear by rubbing. More specifically, a workshop coordinated by the World Health Organization defines leukoplakia as “white plaques of questionable risk having excluded (other) known diseases or disorders that carry no increased risk for cancer” (26). The erythroplakia is a clinical term reserved as an exclusion diagnosis for a red velvety mucosal lesion that cannot be classified otherwise. These are clinical terms based on the macroscopic appearance, the lesion, and not pathologic diagnoses, and inference on the histopathology of the lesion
should not be made. Histologically, most leukoplakia are the consequence of a hyperkeratotic response to an irritant, and only 20% shows evidence of dysplasia or carcinoma on histology (27, 28), while 90% of erythroplakia demonstrate changes consistent with premalignancy or early malignancy (29). However, it is now evident that the macroscopically normal mucosa that surrounds the excision site of HNSCC may also represent mucosa in an inconspicuous state of premalignancy, and may be the source of recurrence (10). As early as the 1950s, the concept of field cancerization that the mucosa is diffusely diseased and at higher risk of developing tumor was proposed (30). Scientific advances over the past 20 years have begun to describe the pathogenesis of HNSCC at a molecular and genetic level.
should not be made. Histologically, most leukoplakia are the consequence of a hyperkeratotic response to an irritant, and only 20% shows evidence of dysplasia or carcinoma on histology (27, 28), while 90% of erythroplakia demonstrate changes consistent with premalignancy or early malignancy (29). However, it is now evident that the macroscopically normal mucosa that surrounds the excision site of HNSCC may also represent mucosa in an inconspicuous state of premalignancy, and may be the source of recurrence (10). As early as the 1950s, the concept of field cancerization that the mucosa is diffusely diseased and at higher risk of developing tumor was proposed (30). Scientific advances over the past 20 years have begun to describe the pathogenesis of HNSCC at a molecular and genetic level.
Experimental animal models of tumor formation have identified chemical tumorigenesis as a two-step process. First, a mutagen, such as dimethylbenzanthracene (DMBA) or 4NQO, is applied to the epithelium (31). During this step, called tumor induction, genotoxic stress induces DNA damage, which, if not repaired, leads to mutation of the genome of a precursor cancer cell. In this model, mutations are not sufficient to induce cancer. The mutated precursor cancer cell needs to be stimulated to proliferate to progress to cancer. In skin carcinogenesis, application of croton’s oil stimulates the protein kinase C (PKC) signaling cascade (see below) and induces epithelial and precursor cancer cell proliferation; this step is called tumor promotion (32). With further mutations, clonal outgrowth becomes irreversible and unregulated.
The critical mutations leading to cancer development occur in two major gene categories: proto-oncogenes and tumor suppressors. Proto-oncogenes are genes involved in normal cellular functions such as cell proliferation or cell death (33). Ras and epidermal growth factor receptor (EGFR) are proto-oncogenes. A proto-oncogene with a mutation that renders it capable of transforming a cell is called an oncogene. Since oncogenic mutations represent genetic gain of function, mutation in only one of the two gene copies (alleles) is sufficient to confer a growth advantage. For example, only one mutated copy of an activated EGFR is sufficient to drive proliferation. In contrast, tumor suppressors are genes that prevent carcinogenesis. Mutations of both alleles are required to remove a normal cellular block to cancer development (33). Often one copy is inactivated by mutation, and the other is lost secondary to loss of heterozygosity (LOH). For instance, p53 is a tumor suppressor, as it prevents cell proliferation and induces apoptosis. With a mutation that inactivates only one copy of p53, the cell can still efficiently regulate the cell cycle via the remaining non-mutated copy. If copies are lost or inactivated, the cell cycle block is removed. Ironically, p53 was initially identified in a screen designed to identify oncogenes (34, 35, 36). The mutant copy of p53 that was thought to be an oncogene was later recognized to be a dominant negative form, in which the mutated p53 prevented the function of the normal allele (37, 38). When this was understood, p53 was recognized to be a tumor suppressor (39, 40). A list of putative and confirmed oncogenes and tumor suppressors in HNSCC and their respective chromosomal locations is provided (Table 108.1).
A multistep model of HNSCC progression proposes that mutations may be acquired in a progenitor cancer cell in a successive manner, ultimately leading to carcinoma formation (Fig. 108.1) (7, 8, 33, 41). This model resulted from the analysis of cancers and cancer precursors in which histology was linked to genetic analysis (42). LOH of chromosome 3p and 9p, which contains the tumor suppressor p16, is frequently found in dysplasias, and is thought to be involved early during carcinogenesis (6, 8). Loss of 17p, the part of the genome that contains p53, occurs at the transition from a noninvasive to an invasive lesion (6). In contrast, alteration of chromosomes 4q, 11q, and 8 is found
preferentially in carcinomas and is thought to represent late events in the carcinogenic process (6, 7). 11q amplification corresponds to cyclin D1 overexpression.
preferentially in carcinomas and is thought to represent late events in the carcinogenic process (6, 7). 11q amplification corresponds to cyclin D1 overexpression.
TABLE 108.1 TUMOR SUPPRESSORS AND ONCOGENES IN HNSCC | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
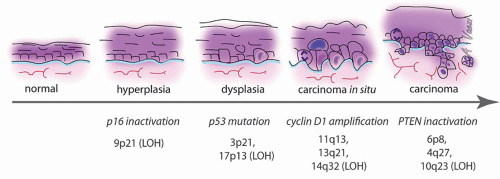 Figure 108.1 Tumor progression in HNSCC. Timeline representing the correlation between histopathology and cytogenetics in HNSCC and its precursor lesions. Histology is represented on the top, cytogenetic abnormalities on the bottom. |
In LOH and chromosomal amplification, a group of genes located in one region of a chromosome are all lost or amplified at once. Genetic mapping of the lost/amplified portion of the chromosome helps delineate genes potentially involved in tumor formation. In addition, cells with common genetic alterations can be identified on histology and mark clonal expansion of cancer progenitors. By labeling cells bearing these chromosomal markers together with p53 mutations on histologic slides, it was found that in 35% of oral carcinomas the macroscopically normal mucosa surrounding the tumor contained genetic alteration (43). Eradication of the mucosa bearing genetic alterations at the time of surgery is critical to reduce local recurrences (44). These data support the field cancerization theory put forward in the 1950s.
With the principles of carcinogenesis in mind, we can probe into the major molecular pathways that are dysregulated in cancer precursor cells and are instrumental in driving cancer progression. The clonal growth of a cancer precursor cell can be viewed as the product of a steady state equation in which the rate of proliferation is greater than the rate of cell death, leading to a net overgrowth of the clone. At the core of proliferation lies the cell cycle and its tight regulation. Before elaborating on this, it is important to review the major autocrine and paracrine signals that drive the cell cycle.
UPSTREAM OF THE CELL CYCLE: EPIDERMAL GROWTH FACTOR RECEPTOR AND TGFβ
Epidermal Growth Factor
Under physiologic conditions, epithelial cells proliferate in response to mitogens of the epidermal growth factor (EGF) family of ligands that are secreted in autocrine or paracrine fashion, or presented via cell-cell contact and activate the EGF receptor (EGFR) (Fig. 108.2A). For instance, cells are stimulated to proliferate via EGF during wound healing. The signal is initiated by 1 of 11 members of the EGF family. All EGF family ligands exist in pro-form as transmembrane precursor proteins. They are converted to an active diffusible form by extracellular cleavage of the membrane protein. Six members of the EGF family are expressed in most SCC cell lines and activate the EGFR: EGF, transforming growth factor alpha (TGFa), HB-EGF, amphiregulin, betacellulin, and heregulin (45). HB-EGF is particularly interesting. The proform may either be cleaved to generate a diffusible factor by matrix metalloproteases (MMPs) that degrade extracellular matrix during cell migration (46), or it may activate EGFR as a transmembrane protein via cell-cell interaction, acting in a juxtacrine fashion. The effects on the target cell are different depending on whether HB-EGF is soluble or membrane bound. (Singh AB, Harris R. Autocrine, paracrine and juxtacrine signaling by EGFR ligands. Cell Signal 2005;10:1183-1193.)
Growth factors present in the interstitial space can also act on other receptors of the EGFR family (also called the ErbB family) that include ErbB1 (EGFR), ErbB2 (HER2/cneu), ErbB3 (Her3), and ErbB4 (Her4) (45). In absence of ligand, most ErbB-family receptors are present at the cell surface of epithelial cells in the form of monomeric inactive transmembrane tyrosine kinase receptors. Binding of bivalent extracellular ligand triggers receptor dimerization, which brings catalytic cytoplasmic tails into close proximity, allowing trans-autophosphorylation of the cytoplasmic tails on tyrosine, serine, and threonine residues (47, 48). ErbB receptors present at the cell surface are not a static pool of proteins. The receptors constantly cycle between an endosomal reservoir and the cell surface, where they become exposed to the ligands (49). After being activated, the receptor is endocytosed and either degraded or recycled (47, 49).
Upon activation and transphosphorylation, active EGFR triggers three major signaling pathways. First, it activates the Ras-Map kinase pathway. Autophosphorylation sites on the cytoplasmic tail become docking sites for the adaptor protein SHC, which in turn recruits Grb2, which recruits Sos. Sos is a guanosine exchange factor for Ras, a small GTPase bound to the inner leaflet of the plasma membrane where it cycles between GDP and GTP bound forms (50, 51). Ras acts as a molecular switch which is turned “on” when bound to GTP (52). The Sos activates Ras by mediating the exchange of GDP for GTP. Active Ras recruits the scaffolding protein Raf to the membrane (53). This leads to activation of the map kinase cascade, ultimately leading to ERK
and Jun kinase (JNK) phosphorylation and activation (54). JNK is a serine/threonine kinase, which phosphorylates the c-Jun transcription factor. Phosphorylated c-Jun forms a heterodimer with phosphorylated c-fos, which together constitutes the AP-1 early response transcription factor that drives cell proliferation (55). ERK can also phosphorylate and activate transcription factors responsible for cell proliferation and cell migration.
and Jun kinase (JNK) phosphorylation and activation (54). JNK is a serine/threonine kinase, which phosphorylates the c-Jun transcription factor. Phosphorylated c-Jun forms a heterodimer with phosphorylated c-fos, which together constitutes the AP-1 early response transcription factor that drives cell proliferation (55). ERK can also phosphorylate and activate transcription factors responsible for cell proliferation and cell migration.
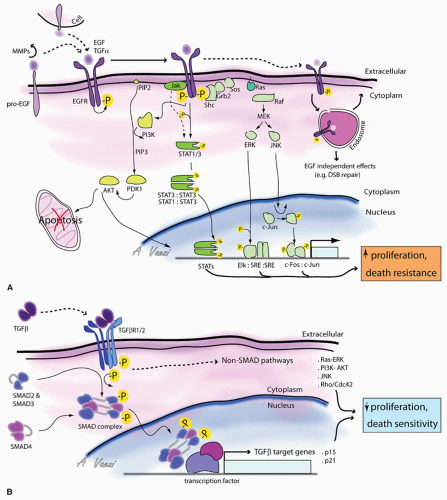 Figure 108.2 Signaling upstream of the cell cycle. Model for EGFR (A) and TGF-β (B) signaling pathways are represented. See text for details. |
The second signaling pathway activated by EGFR is the Jak/STAT pathway. Inactive STAT (signal transducer and activator of transcription) proteins are located in the cytoplasm. Activated EGFR recruits and activates the transcription factor STAT3 via two mechanisms: via Janus kinase (Jak) or via direct interaction with STAT3. Jak is a tyrosine kinase that binds to the juxtamembrane intracellular portion of activated receptors. Active Jak then phosphorylates
and activates STAT1 or STAT3 proteins (56). An alternate mode of activation of STAT3 is via direct interaction with EGFR (57). STAT3 is recruited to active EGFR, where it binds to specific phosphotyrosine residues of the receptor via its Src homology domain (SH2). STAT3 is then directly phosphorylated by the receptor on critical tyrosine and serine residues. Phosphorylation of STAT3, either via Jak or via EGFR, leads to its dimerization, either as STAT3 homodimers or as STAT3:STAT1 heterodimers (58). Activated STAT transcription factor dimers translocate either to mitochondria where they regulate metabolism characteristic of cancer cells (59, 60) or to the nucleus where they regulate growth factor/cytokine-directed gene expression (56) and increase cell motility (61, 62).
and activates STAT1 or STAT3 proteins (56). An alternate mode of activation of STAT3 is via direct interaction with EGFR (57). STAT3 is recruited to active EGFR, where it binds to specific phosphotyrosine residues of the receptor via its Src homology domain (SH2). STAT3 is then directly phosphorylated by the receptor on critical tyrosine and serine residues. Phosphorylation of STAT3, either via Jak or via EGFR, leads to its dimerization, either as STAT3 homodimers or as STAT3:STAT1 heterodimers (58). Activated STAT transcription factor dimers translocate either to mitochondria where they regulate metabolism characteristic of cancer cells (59, 60) or to the nucleus where they regulate growth factor/cytokine-directed gene expression (56) and increase cell motility (61, 62).
Finally, EGFR also activates the PI3 kinase/Akt pathway that promotes cell survival by suppressing apoptosis. Activated EGFR recruits and activates phospholipase-Cγ, which directs the hydrolysis of the phosphatidylinositol biphosphate (PIP2) at the plasma membrane. This hydrolysis generates an insoluble diacyl glycerol, which activates PKC, and a soluble phosphatidyl (1, 4, 5) triphosphate in the cytosol (32). There, IP3 (1, 4, 5) releases Ca2+ stores from the endoplasmic reticulum. Activated EGFR can also recruit and activate PI3 kinase, via the SH2 domain of the kinase. PI3 kinase then phosphorylates PIP2 to generate phosphatidylinositol-(3, 4, 5) triphosphate (PIP3) that remains membrane bound (63). Proteins with a pleckstrin homology (PH) domain such as Akt and PDK migrate to and associate with PIP3 located at the plasma membrane (63). Close proximity of Akt and PDK leads to Akt phosphorylation and activation. Akt renders the cell more resistant to apoptosis and increases proliferation. In addition, Akt also induces a motile and invasive cell phenotype. PTEN, an important tumor suppressor that is frequently mutated in cancers, counteracts Akt effects (64, 65). PTEN’s sequence resembles serine, threonine, and tyrosine phosphatases, but it acts principally as a lipid phosphatase converting PIP3 into PIP2 (64). Thus, PTEN tempers the effect of PI3K activation by limiting PIP3-mediated signaling. PI3 kinase lipid signaling pathway is a major pathway linking EGFR activation to increased cell proliferation and motility and decreased cell death, and is often involved in carcinogenesis.
Clinical Implications of EGF Pathway in HNSCC
EGFR is frequently upregulated in HNSCC (66, 67, 68), and in general EGFR upregulation correlates with a worse prognosis (68, 69, 70), including more rapid disease progression and the presence of lymphatic metastasis, through its effect on proliferation, motility, and angiogenesis (71). The mechanisms leading to EGFR overexpression in HNSCC are still unclear. Genome sequencing suggests that EGFR is not mutated in HNSCC, in contrast to other tumor types (3). It is rarely amplified (less than 10% of HNSCC). However, a peculiarity of HNSCC is the common overexpression of a truncated mutant of the receptor (EGFRvIII) in 40% of HNSCC (72). EGFRvIII, which is constitutively active and has degradation prolonged half-life, is found exclusively in tumors, making it a prime target for anticancer therapy with specific monoclonal antibodies. Because of the high frequency of EGFR upregulation HNSCC, EGFR-targeting strategies have been extensively investigated. Both small molecule EGFR tyrosine kinase inhibitors and monoclonal antibodies have been tested for activity against HNSCC. Cetuximab, an EGFR-blocking antibody, demonstrated activity against HNSCC, with improved locoregional control and survival compared to radiotherapy alone (73) and with improved response rate and survival when used in combination chemotherapy (74). Cetuximab (Erbitux) is now used routinely to potentiate radiotherapy. To date, while tyrosine kinase inhibitors targeting EGFR (such as gefitinib and erlotinib) have activity in HNSCC, they have not demonstrated prolonged survival in Phase III clinical studies (75). This discrepancy suggests that EGFR-blocking antibody has effects other than inhibition of the signaling pathway; the antibody could have an effect on immunity (76, 77), or on EGF-independent effects such as DNA repair (78, 79).
In addition to targeting EGFR itself, considerable effort has been invested in inhibiting downstream targets. STAT3 homodimers, which have oncogenic properties, are overexpressed in HNSCC and are the target of anticancer therapies (80). Specifically, STAT3 inhibitory strategies using dimerization-disrupting agents or competitive inhibitors of the DNA-binding domain of STAT3 have been proposed to combat HNSCC (80, 81, 82); the latter was used in a clinical trial with promising results. HNSCC genome sequencing found that 5% of tumors display H-Ras mutations, leading to constitutive activation of the small GTPase (3). Whether Ras is mutated or simply subject to sustained activation by EGFR, it is a seductive therapeutic target. Ras may be inhibited via blocking farnesylation of the protein (63), an essential posttranslational modification required for its targeting to the plasma membrane where it mediates its effects. In addition, the PIP3 lipid pathway is also relatively frequently mutated in HNSCC. PTEN is mutated in 7% of HNSCC with some cancers bearing both H-Ras and PTEN mutations (3, 83). The inactivation of PTEN promotes PIP3 accumulation and activation of downstream targets such as Akt. PI3KCA, a catalytic subunit of PI3 kinase, is also mutated in HNSCC (3, 84). These mutations underscore how frequently the inositol lipid pathway is the target of carcinogenic mutations. While the contribution of these mutations to protein function or prognosis is yet unknown, they will likely be the subject of exciting HNSCC tumor biology discoveries in the near future. To date, therapeutic strategies targeting the PI3K/Akt pathway in HNSCC remain mostly limited to the preclinical setting (63). Pharmacologic inhibitors being developed include a water-soluble form of wortmannin, an inhibitor of PI3K.
Importantly, mTOR, a downstream target of Akt, can be inhibited with rapamycin, the RAD001(Everolimus), and CCI-779 (Temsirolimus) compounds and is the focus of innovative anti-HNSCC therapeutic strategies that are already being tested in Phase I and II clinical trials (85).
Importantly, mTOR, a downstream target of Akt, can be inhibited with rapamycin, the RAD001(Everolimus), and CCI-779 (Temsirolimus) compounds and is the focus of innovative anti-HNSCC therapeutic strategies that are already being tested in Phase I and II clinical trials (85).
Transforming Growth Factor Beta
The transforming growth factor beta (TGF-β) superfamily of ligands regulates both embryologic processes and adult tissue homeostasis. These ligands act as tumor suppressors by inhibiting proliferation of epithelial cells, and by sensitizing cells to apoptosis; in that respect, TGF-β effects are opposite to those of EGFR. It is therefore not surprising that TGF-β signaling is also the target of carcinogenic mutations.
The TGF-β superfamily includes TGF-βs (TGF-β 1, 2, and 3), bone morphogenic protein, growth and differentiation factors, nodal, activin (involved in embryogenesis and bone formation), and anti-mullerian hormone (86, 87). TGF-β is secreted as inactive precursors that are proteolytically activated by extracellular proteinases (86, 88). The biologically active cytokine is present in the interstitial compartment; the signal is triggered when TGF-β binds a dimeric Type II TGFβ receptor. TGF-β receptors are single-span transmembrane receptors with serine/threonine kinase activity (Fig. 108.2B). Upon activation, dimeric type II receptors recruit and associate with dimeric Type I receptors, another transmembrane serine/threonine kinase receptor, to form a tetrameric receptor complex (86, 87, 88). Both receptors are then in close proximity, leading to the phosphorylation and activation of the Type I receptor by the Type II receptor. In turn, activated Type I receptor recruits an adaptor protein named SARA (Smad anchor for receptor activation) that mediates transient binding between the Type I receptor and SMADs, a family of transcription factors present in the cytoplasm. The SMAD family of proteins has eight members, SMAD 1-8, which come in three types: receptor-activated SMADS (R-SMADS), co-SMADS, and inhibitory SMADS (I-SMADS) (89). R-SMADS involved in TGF-β signaling are SMAD2 and SMAD3. SARA brings SMAD 2 and 3 in close proximity to the catalytic site of the Type I receptor, which then phosphorylates the R-SMAD C-terminus (87, 88). This posttranslational modification induces a conformational change that allows interaction with a co-SMAD (SMAD4). The newly assembled SMAD:co-SMAD complex translocates to the nucleus where it interacts with other transcription regulators, to control expression of genes involved in apoptosis, and cell cycle repressors including p15 and p21(87). However, TGF-β signaling does not necessarily involve SMADs, and SMADs are not necessarily activated by Type I receptors. Recently, important cross talk between the MAP kinase pathway and SMADs has been revealed: ERK has been shown to phosphorylate SMADS (90). Reciprocally, TGF receptors can regulate important targets in a SMAD-independent manner, including the Map kinase pathway (90) and the Rho family of small GTPases (91), which are “molecular switches” similar to Ras, but are involved in the regulation of actin dynamics, cell migration, and metastasis (51). This represents one way in which TGF-β regulates cell morphology and migration.
The TGF-β pathway is negatively regulated at three levels. First, at the level of the extracellular matrix, the small proteoglycan decorin inhibits TGF-β receptor signaling (92). Decorin has antitumoral activity in vitro. Second, intracellular I-SMADs prevent binding of R-SMADs to the activated Type I receptor, and thus negatively impact SMAD signaling. Third, both the receptor and SMADs can be targeted for proteasome-mediated degradation, regulated by E3 ubiquitin ligases SMURF1 and SMURF2 (93, 94). Interestingly, SMURF1 not only downregulates the TGF-β signaling pathway but also enhances cancer cell migration and invasion by targeting the GTPase Rho A for degradation, thereby reducing stress fibers and enhancing cell protrusions and motility (91). In summary, TGF-β signaling is at the crossroads of cell proliferation, cell death, and migration, and it is involved in many aspects of carcinogenesis.
Clinical Implications of TGF-β Pathway in HNSCC
The role of TGF-β signaling in tumorigenesis is multifaceted. While TGF-β has tumor suppressive effects, cancer cells can circumvent the suppressive component of the pathway and use TGF-β stimulation to their advantage to promote invasion and secretion of mitogens and pro-metastatic cytokines. This explains why TGF-β signaling is inhibited in certain cancers and upregulated in others. HNSCC is no exception. For instance, TGF-β receptor and SMADS are often downregulated in HNSCC (95). The downregulation may occur via loss of heterozygosity of chromosome q18, which contains TGFβRI and SMAD genes, or via DNA methylation-mediated gene repression (88). In addition, inactivating mutations in TGFβRI or II and SMADs have been reported in HNSCC, and may be present in up to 20% of neck metastasis (TGFβRI) (96, 97, 98). However, exosome sequencing of 80 HNSCC primary tumors found no mutations in TGF-β pathway, indicating that inactivating mutations are probably not a frequent event (3). An elegant demonstration that the downregulation of TGF-β signaling participates in HNSCC formation comes from a genetic mouse model in which the loss of TGFβRII, in the presence of an activating Ras mutation, induces head and neck tumors that closely resemble human HNSCC.
As above, TGF-β1 is actually frequently overexpressed in HNSCC, by about five times the normal levels in 40% of HNSCC (99). TGF-β1 is not only upregulated in the tumor but also in the surrounding normal-appearing tissue, suggesting that the tumor microenvironment contributes to tumorigenesis. This hypothesis was confirmed in a
transgenic animal model where the induction of TGF-β1 overexpression in oral mucosa led to mucosal hyperproliferation and angiogenesis (99). This suggests that TGF-βR ligand stimulation may be upregulated in HNSCC and have a profound effect on tumorigenesis and tumor biology. In addition, modulation of TGF-β signaling mediators downstream of the receptor also contributes to HNSCC tumorigenesis. For instance, SMAD2 and 4, but not SMAD3, are frequently downregulated in HNSCC, correlating with a worse prognosis (100, 101). Here again, a genetic animal model in which SMAD4 is deleted demonstrates that loss of SMAD4 interferes with DNA repair pathways and results in the spontaneous development of invasive head and neck tumors (101). In a similar vein, TGF-β inhibitors interfering with SMAD signaling also contribute to tumorigenesis. The overexpression of SMURF2, which enhances cell migration and invasion in vitro, is a marker of bad prognosis in esophageal SCC (102); whether SMURFs have prognostic value in HSNCC has not yet been tested.
transgenic animal model where the induction of TGF-β1 overexpression in oral mucosa led to mucosal hyperproliferation and angiogenesis (99). This suggests that TGF-βR ligand stimulation may be upregulated in HNSCC and have a profound effect on tumorigenesis and tumor biology. In addition, modulation of TGF-β signaling mediators downstream of the receptor also contributes to HNSCC tumorigenesis. For instance, SMAD2 and 4, but not SMAD3, are frequently downregulated in HNSCC, correlating with a worse prognosis (100, 101). Here again, a genetic animal model in which SMAD4 is deleted demonstrates that loss of SMAD4 interferes with DNA repair pathways and results in the spontaneous development of invasive head and neck tumors (101). In a similar vein, TGF-β inhibitors interfering with SMAD signaling also contribute to tumorigenesis. The overexpression of SMURF2, which enhances cell migration and invasion in vitro, is a marker of bad prognosis in esophageal SCC (102); whether SMURFs have prognostic value in HSNCC has not yet been tested.
THE CELL CYCLE ORCHESTRATES CELL PROLIFERATION
Multiple extracellular mitogenic signals such as EGF, the sensing of neighboring cells via cell-cell interactions, or tissue oxygenation are integrated via distinct signaling pathways that converge to regulate the cell cycle. The integration of these signals informs the cell of the decision to proliferate. The cell cycle-regulating machinery is a prime target for oncogenic mutations, since cancer is a disease of unregulated cell proliferation.
In the resting state, the cell is quiescent, termed the G0 phase of the cell cycle. In this dormant stage, the pocket family proteins (Rb, p107, and p130) act as transcriptional cofactors that inhibit expression of proliferationrelated genes (103). Among other effects, Rb inhibits the transcription factor E2F that drives the expression of proliferation genes (33, 103). However, in the presence of growth factors, the Ras/Raf/ERK pathway becomes activated, leading to the phosphorylation and activation of transcription factors driving expression of proliferation genes, including cyclin D (Fig. 108.3) (104). Cyclin proteins are expressed for a limited period during the cell cycle and are rapidly degraded via ubiquitin-mediated protein degradation, leading to oscillating concentration during the cell cycle (105). There are several members of the cyclin family, each specific for a phase of the cell cycle. Cyclins have no intrinsic enzymatic activity, but associate with and partially activate cyclin-dependent kinases (Cdk), whose expression levels remain constant during the cell cycle. A second posttranslational modification, such as phosphorylation of the cyclin:Cdk complex by a Cdk-activating kinase (CAK or cylinH:Cdk7) at a specific threonine site (Thr 160) (106) or dephosphorylation of an active site by cdc25 (107), is also required for full activity of the complex.
Early in the G1 phase, cyclin D is synthesized as a consequence of ERK activation (104). Cyclin D accumulates as long as the growth factor is present, but in the absence of growth stimuli, the protein is quickly degraded, and progression through the cell cycle stops. Cyclin D associates with Cdk4/6, the effector kinases of the G1 phase of the cell cycle. Active cyclinD:Cdk4/6 complex acts in a positive feedback loop during the G1 phase to promote cyclin D expression. The active kinase complex mediates the inhibitory phosphorylation of pocket proteins, which repress E2F.
As a result, E2F-dependent transcription is derepressed, and cyclin D synthesis increases (104). The same mechanism is responsible for the accumulation of cyclin E later in G1. As cyclin E levels increase in somatic cells, it associates with Cdk2 to form an active complex that governs the G1/S transition. This cyclin:Cdk complex has distinct functions to prepare cell for DNA synthesis and mitosis. First, it induces duplication of the centrosome (108), a membraneless organelle responsible for microtubule network and mitotic spindle assembly. Second, it renders the cell competent for DNA synthesis, but does not start DNA synthesis (109). Third, it phosphorylates and inactivates a cell cycle inhibitor named p27 (110). The transcription of cyclin A thus far inhibited by p27 is now induced. Cyclin E is present at high levels only at the G1/S transition in somatic cells and is targeted for ubiquitin-mediated degradation in early S phase. As cyclin E levels decrease, the free pool of Cdk2 instead associates with the increasingly abundant cyclin A. The active cyclin A:Cdk2 then stimulates DNA replication by activating the replication complex, completing the transition from G1 to S phase (104, 109).
After completion of DNA replication, cyclin A changes kinase partner, now associating with Cdk1, which drives the G2 and M phases. G2 is a phase of preparation for mitosis that is incompletely understood. G2 is not an absolute requirement for division, as cancer cells may skip this stage and directly progress to mitosis. In early G2, cyclin B accumulates. As the cell progresses through G2, cyclin A is targeted for ubiquitin-mediated degradation, increasing the pool of free Cdk1 available for association with cyclin B. With rising cyclin B:Cdk1 activity by the end of G2, nuclear breakdown occurs, chromosomes condense, and the cell enters mitosis (111).
Mitosis is a highly orchestrated phenomenon. During this phase, 46 duplicated chromosomes need to be distributed equally between two future daughter cells. Faulty distribution results in aneuploidy, which is most often incompatible with cell survival, at least under physiologic conditions (112). The central regulator of proper cell division, ensuring correct distribution of genetic material between daughter cells, is an ubiquitin ligase complex called the anaphasepromoting complex (APC). APC targets mitosis regulatory proteins for degradation. The specificity of APC for its targets is dependent on Cdc20; if Cdc20 is inhibited, APC cannot degrade regulators essential for progression through mitosis, and the cell cycle is stalled before anaphase (105).
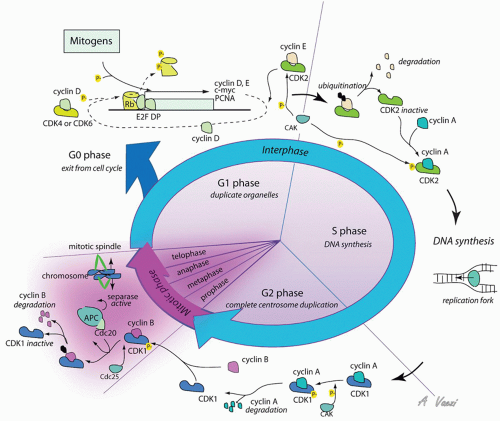 Figure 108.3 Cell cycle proteins regulate cell replication. Cyclin:Cdk complexes orchestrate progression through the cell cycle. Mitogen signals triggering proliferation are represented on the top left; regulation of cyclin:Cdk complexes is represented on the rest of the figure. See text for details. |
During mitosis, chromosomes condense, the nuclear envelope breaks down, a bipolar mitotic spindle forms, and each chromosome attaches to the mitotic spindle at the chromosome center, or kinetochore. To ensure symmetry, the “mitotic checkpoint” ensures that no cell division starts before each and every chromosome is attached to both mitotic spindles (113). The molecular mechanism responsible for preventing premature division is only partially understood. Uncaptured kinetochores bind to protein complexes formed by Bub and Mad proteins (114, 115




Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
