THE CHILDHOOD GLAUCOMAS constitute a rare, heterogeneous, and vision-threatening group of disorders. Pediatricians and eye care providers are often the first health care professionals to encounter children with glaucoma. Familiarity with the clinical features of this disease, and with the children most at risk for developing glaucoma, may increase correct diagnosis and timely treatment for affected children. In adults, glaucoma is often occult; however, in children, strong suggestive signs of glaucoma are present more often. While there is considerable common ground, children with glaucoma often require examination techniques and treatment strategies, which differ markedly from those most appropriate for their adult counterparts. Genetic, pharmacologic, and technologic advances in the diagnosis and treatment of glaucoma raise the hope that this disease will one day (some day) no longer cause visual impairment in either adults or children.
Many classification systems have been proposed for the childhood glaucomas, and most subdivide them into those of primary and secondary origin (1). Hence, a primary glaucoma is one caused by an intrinsic disease of the aqueous outflow mechanism and is often of genetic origin, whereas a secondary glaucoma is one caused by another ocular disease, injury, drug, or systemic disease (Table 12.1). Both primary and secondary pediatric glaucomas may be associated with significant systemic conditions. It is therefore important for the ophthalmologist to accurately interpret eye signs as clues for the diagnosis and classification of both the glaucoma and associated systemic disease (2). Continued elucidation of the genetic underpinnings of many childhood glaucomas may someday facilitate a classification relying more on underlying genetic abnormalities rather than phenotypically driven diagnostic labels.
SIGNS AND SYMPTOMS OF GLAUCOMA IN CHILDREN
The signs and symptoms of glaucoma vary greatly among children, according to the age of the child and the suddenness and severity of the intraocular pressure (IOP) elevation. During the first year of life, glaucoma is commonly suspected because of signs and symptoms related to secondary corneal changes. Older children are seen more often with loss of vision from chronic glaucoma or with symptoms of pain and vomiting related to acute glaucoma. Elevation of IOP is required to confirm the diagnosis of childhood glaucoma, although its presence (past or present) may be strongly suspected on the basis of classic symptoms and other signs of the disease (see below). While somewhat lower in young infants than in school age children and adults, the range of normal IOP in childhood approximates the normal adult range; rarely are normal measurements above 22 mmHg or below 10 mmHg. Accurate IOP measurements are essential not only to the diagnosis but also to the management of children with glaucoma (see later under Examination).
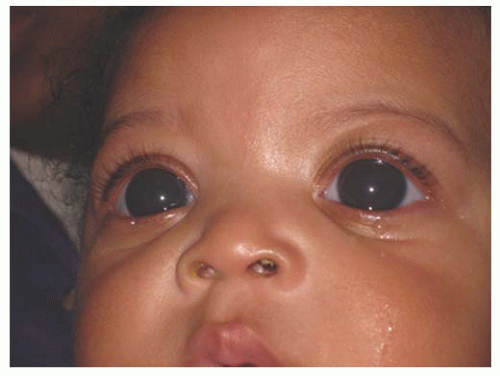
Infants and young children with glaucoma usually present for ophthalmologic evaluation because the pediatrician or parents have noted something unusual about the appearance of the child’s eyes or behavior. Corneal opacification and/or enlargement (a response to elevated IOP) are the signs most commonly heralding glaucoma in the infant; both may progress over the first 2 years of life if IOP remains elevated (Figs. 12.1 and 12.2). At other times, the child’s glaucoma may manifest itself as one or more of the “classic triad” of findings: epiphora, photophobia, and blepharospasm (Fig. 12.3) (3). Photophobia and epiphora result from corneal edema (often with associated breaks in Descemet membrane called Haab Striae). The baby may be noted to withdraw from light or to bury his head against his parent or bedding to prevent exposure to light; eye rubbing may also be noted. Even indoors, the infant may show an apparent reluctance to face upward, and may mistakenly be considered shy (Fig. 12.4).
These corneal signs and symptoms of glaucoma in early life may be of sudden onset, with dramatic opacification of the cornea and onset of photophobia occurring over a few hours. Such an acute onset of signs is probably related to initial or additional breaks in Descemet membrane (Figs. 12.5 and 12.6). The occurrence of breaks appears to be confined to the first 2 years of life, when rapid expansion of the cornea may occur secondary to glaucoma. Breaks are permanent and remain as important evidence of early glaucoma; although some are subtle, at other times significant associated corneal scarring may result (Fig. 12.7). The defects appear as wavy parallel lines on the inner side of the cornea and are usually curvilinear and horizontal. They represent the separated edges of Descemet membrane. Breaks with more vertical orientation may be seen secondary to the acute bending of the cornea rarely occurring with delivery using forceps (Fig. 12.8) (4).
Table 12.1 PRIMARY AND SECONDARY CHILDHOOD GLAUCOMAS
B. Autosomal dominant juvenile (open-angle) glaucoma
C. Primary angle-closure glaucoma
D. Associated with ocular abnormalities
Iridodysgenesis
Aniridia
Congenital iris ectropion syndrome
Iridotrabecular dysgenesis (iris hypoplasia)
Corneodysgenesis (or iridocorneal dysgenesis)
Axenfeld-Rieger anomaly
Peters anomaly/syndrome
Congenital microcornea with myopia
Sclerocornea
Congenital hereditary endothelial dystrophy
Posterior polymorphous dystrophy
Anterior corneal staphyloma
Iridocorneal endothelial syndrome
Other
Idiopathic or familial elevated episcleral venous pressure
E. Associated with systemic abnormalities
Chromosomal abnormalities:
Trisomy 13 (Patau syndrome)
Trisomy 18 (Edward syndrome)
Trisomy 21 (Down syndrome)
Turner syndrome (XO)
Connective tissue abnormalities:
Stickler syndrome
Marfan syndrome
Metabolic disorders:
Hepatocerebrorenal syndrome
Oculocerebrorenal (Lowe) syndrome
Mucopolysaccharidosis
Phacomatoses:
Sturge-Weber syndrome
Neurofibromatosis Type 1 (NF-1)
Nevus of Ota (congenital ocular melanosis)
von-Hippel-Lindau syndrome
Other:
Rieger syndrome (Axenfeld-Rieger syndrome)
SHORT syndrome
Rubinstein-Taybi syndrome
Infantile glaucoma with mental retardation and paralysis
Oculodentodigital dysplasia
Open-angle glaucoma associated with microcornea and absence of frontal sinuses
Caudal regression syndrome
Cutis marmorata telangiectasia congenita
Warburg syndrome
Kniest syndrome (skeletal dysplasia)
Michel syndrome
Nonprogressive hemiatrophy
PHACE syndrome
Soto syndrome
Linear scleroderma
GAPO syndrome
Roberts pseudothalidomide syndrome
Wolf-Hirschhorn (4p-) syndrome
Rainbow syndrome
Nail-patella syndrome
Proteus syndrome
Fetal hydantoin syndrome
Cranio-cerebello-cardiac (3C) syndrome
Brachmann-deLange syndrome
Hallerman-Streiff syndrome
II. Secondary glaucomas
A. Traumatic glaucoma
Acute glaucoma
Angle concussion
Hyphema
Ghost cell glaucoma
Late-onset glaucoma with angle recession
Arteriovenous fistula
B. Secondary to intraocular neoplasm
Retinoblastoma
Juvenile xanthogranuloma
Leukemia
Melanoma
Melanocytoma
Iris rhabdomyosarcoma
Aggressive nevi of the iris
C. Secondary to uveitis
Open-angle glaucoma
Angle-blockage glaucoma
Synechial angle closure
Iris bombe with pupillary block
Trabecular endothelialization
D. Lens-induced glaucoma
Subluxation-dislocation and pupillary block
Marfan syndrome
Homocystinuria
Weill-Marchesani
Spherophakia and pupillary block
Phacolytic glaucoma
E. Following surgery for congenital cataract (Aphakic)
Lens tissue trabecular obstruction
Pupillary block
Chronic open-angle glaucoma associated with angle abnormalities
F. Steroid-induced glaucoma
G. Secondary to rubeosis
Retinoblastoma
Coats disease
Medulloepithelioma
Familial exudative vitreoretinopathy
Chronic retinal detachment
H. Secondary angle-closure glaucoma
Retinopathy of prematurity
Microphthalmos
Nanophthalmos
Retinoblastoma
Persistent hyperplastic primary vitreous
Congenital pupillary iris-lens membrane
Topiramate use
Ciliary body cysts
I. Malignant glaucoma (aqueous misdirection)
J. Glaucoma associated with increased venous pressure
Cavernous or dural-venous fistula
Orbital disease
K. Secondary to maternal rubella
L. Secondary to intraocular infection
Acute recurrent toxoplasmosis
Acute herpetic iritis
Endogenous endopthalmitis
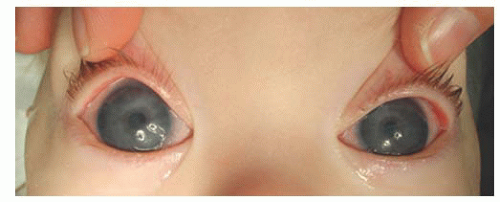
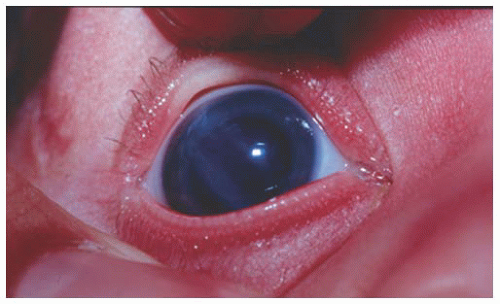
FIGURE 12.1. Corneal enlargement bilaterally, with corneal haze. This infant was 11 months old and had persistent severe corneal enlargement (corneal diameters 14.5 mm both eyes), and corneal edema and opacity with multiple Haab Striae in both eyes. Intraocular pressures were 40 mmHg under anesthesia, on topical medical therapy. Glaucoma was diagnosed at 2 months of age but inadequately treated.
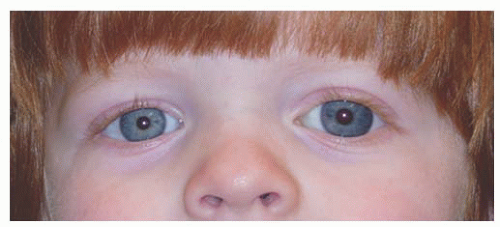
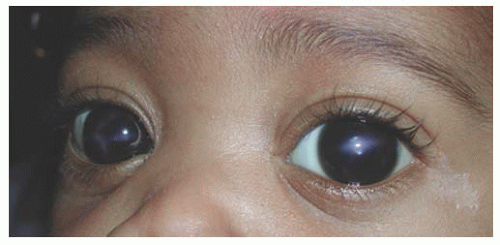
FIGURE 12.2. Corneal enlargement of the left eye that developed slowly over a period of 1 year. Primary infantile glaucoma responded well to angle surgery and medical therapy, and vision improved with glasses for unilateral myopia and patching.
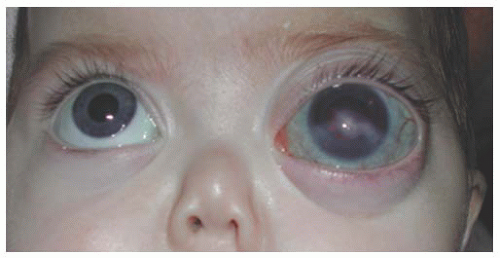
FIGURE 12.3. Tearing of both the eyes caused by uncontrolled primary infantile glaucoma. Note the increased corneal diameter and corneal haze of both eyes.
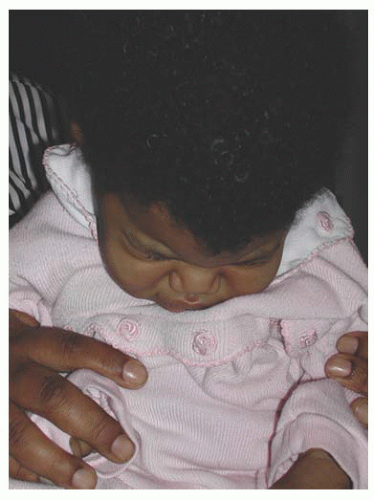
FIGURE 12.4. Hiding of the face caused by photophobia secondary to congenital glaucoma.
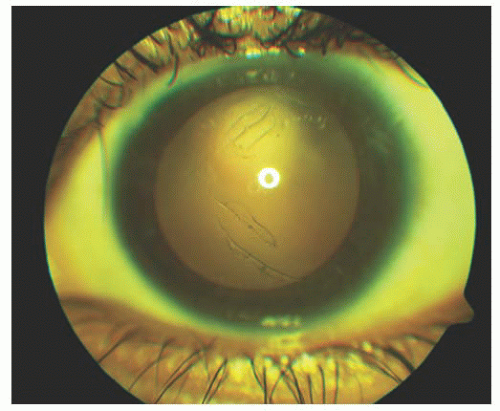
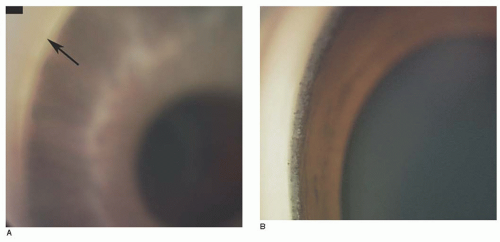
FIGURE 12.5. Haab Striae are seen in retroillumination in the left eye of a teenage girl with advanced primary congenital glaucoma. These marks persist despite her controlled intraocular pressure and resolution of associated corneal edema.
If glaucoma in infancy and early childhood is not treated, progressive enlargement of the cornea may occur throughout the first 2 years of life. The corneal diameter may in extreme cases enlarge to 17 to 18 mm; in these cases, concordant enlargement of the ciliary ring often results in iridodenesis and lens subluxation (Fig. 12.9). Stretched, buphthalmic eyes are easily traumatized, even to the point of rupture.
FIGURE 12.6. Haab Striae seen through a Koeppe lens in a child with congenital glaucoma. Note the curvilinear shape to the break.
FIGURE 12.7. Corneal scarring in a striking pattern, correlating with the presence of Haab Striae. The associated corneal edema has cleared, and the pressure is normalized after goniotomy surgery, but the visually significant corneal opacities remain in both eyes.
FIGURE 12.8. Forceps injury to the right cornea at birth resulted in two parallel linear breaks in Descemet membrane running in an oblique direction across the visual axis; this eye showed high astigmatism and best corrected vision of 20/80, despite aggressive patching.
FIGURE 12.9. Hugely enlarged, buphthalmic left eye, in a 6 month old infant with Pierre Robin syndrome and uncontrolled, congenital glaucoma in that eye. Corneal scarring has resulted from exposure, corneal diameter is 17 mm on the left, and spontaneous posterior lens dislocation has occurred. The eye is blind.
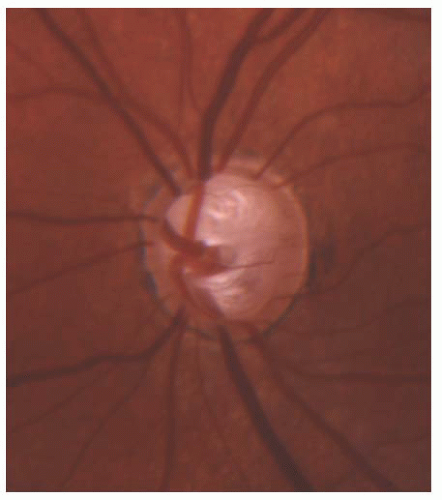
FIGURE 12.10. Advanced cupping in the left eye caused by congenital glaucoma in a 4-year-old girl; vision is preserved at 20/25.
Additional nonspecific signs of glaucoma in early life include the presence of a deep anterior chamber and optic nerve cupping. The extent of optic nerve cupping does not always correlate closely with the anterior segment signs of glaucoma. In the absence of optic atrophy, the optic cup may decrease greatly in size with IOP reduction, and will enlarge again if control of IOP is lost. By contrast, the optic atrophy that may result from chronic or severe IOP elevation is irreversible (Fig. 12.10).
In older children, the anterior segment signs of glaucoma play a less important role in the recognition of this disorder. Of greater importance is the evaluation of the eyes because of decreased vision (usually from induced myopia) or circumstances in which secondary glaucoma might be suspected, such as chronic iridocyclitis, blunt trauma to the anterior segment, neoplasm, or as a consequence of surgery. The presence or absence of optic nerve head cupping in older children is by itself an unreliable diagnostic sign, but remains very important in the follow-up of children with known glaucoma. Older children infrequently present with acute glaucoma inducing nauseating eye pain, headaches, and even colored haloes around lights. This sudden-onset glaucoma may be the result of traumatic hyphema or of angle-closure glaucoma from lens dislocation or cicatricial retinopathy of prematurity. Less frequently, acute glaucoma develops secondary to other processes (Table 12.1).
Loss of vision from childhood glaucoma occurs secondary to pathologic changes in the eye such as corneal opacification and optic nerve damage. Amblyopia is also an important cause of unilateral vision loss, especially in cases where corneal abnormalities and refractive errors are asymmetric.
OCULAR EXAMINATION
The examination of a child with suspected glaucoma includes the components of the complete pediatric eye examination. However, the glaucoma evaluation should address several specific objectives: (a) confirming or excluding the diagnosis of glaucoma, (b) determining the etiology of the glaucoma (if present), and (c) obtaining additional information (including any prior glaucoma treatment) needed to plan for optimal management. If one can confidently exclude the diagnosis of glaucoma, or if an older child with glaucoma is to undergo a trial of medical therapy, examination under anesthesia may not be indicated. If indicated, the anesthetized exam allows more detailed gonioscopy and optic nerve head examination, followed by any indicated surgical treatment.
Vision testing techniques vary greatly with the age of the patient. In infants, good fixation and following and the absence of nystagmus are important indicators of good visual function. In children over 3 years of age, visual acuity and eventually visual field testing can also be assessed.
The external examination is important to detect evidence of associated abnormalities, inflammation, or lacrimal duct obstruction.
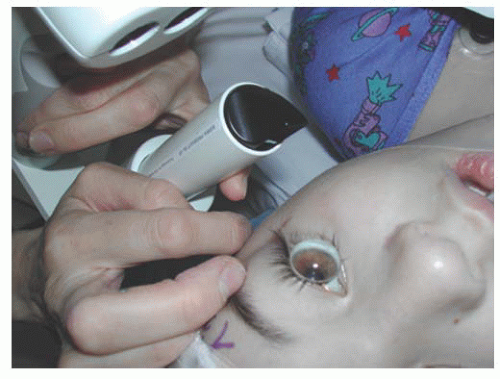
Tonometry should be performed in both the office and operating room settings. Careful measurements of IOP in the office, uninfluenced by general anesthesia, aid in both diagnosing glaucoma and evaluating results of treatment. Among various instruments used to measure IOP in children, the Perkins applanation tonometer (Haag-Streit, Mason, OH) and the Tono-Pen (a hand-held Mackay-Marg-type tonometer, Reichert, Inc, Depew, CA) have been invaluable (5,6), but require the instillation of local anesthetic, which can be anxiety provoking in children. The Icare® rebound tonometer (Finland, Oy), a hand-held tonometer that does not require the use of anesthetic, has demonstrated good clinical correlation when compared with Goldmann applanation among children with glaucoma or suspected glaucoma, and holds promise as an instrument suitable for home tonometry in selected pediatric glaucoma cases (7,8). In older, cooperative children, the standard slit-lamp-mounted Goldmann applanation instrument is often successful.
IOP measurements are variably lowered by sedatives, narcotics, and inhalation anesthetic agents (9,10,11), and variably raised by endotracheal intubation and ketamine (3,12). With the possible exception of chloral hydrate conscious sedation (13), most sedative/anesthetic drugs may alter measured IOP but do not usually normalize very high or asymmetrically elevated IOPs. IOP in normal eyes rises from infancy to reach normal adult levels by middle childhood(14). The range of normal IOP in childhood can be considered from 10 to 22 mmHg (3). Newborns with glaucoma may demonstrate a transient postnatal interval of normal IOP, after which the IOP will again rise.
Careful inspection of the anterior segment provides vital information about a childhood glaucoma patient. The cornea is inspected for changes secondary to elevated IOP. The normal horizontal corneal diameter at birth ranges from 9.5 to 10.5 mm (mean 10 mm), enlarging to approximately 11.5 mm by the end of the second year. Under 1 year of age, diameters of 12 to 12.5 mm are suggestive of glaucoma, and a measurement of 13 mm or more at any time in childhood strongly suggests abnormality, as does asymmetry in corneal diameter between the two eyes of a child (3,15,16,17).
The depth and clarity of the anterior chamber are assessed. The pupil and irides are examined for evidence of primary anomalies or abnormalities secondary to other eye diseases (e.g., aniridia, Axenfeld-Rieger syndrome, and ectropion uveae).
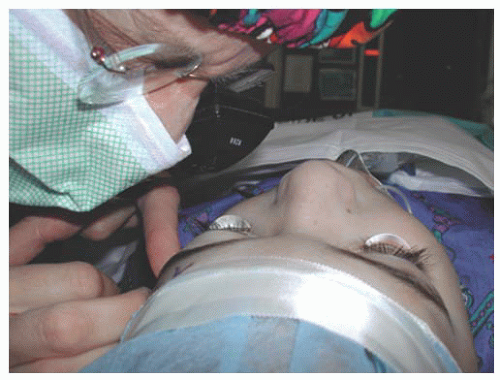
Gonioscopy provides the most important anatomic information about the mechanism of the glaucoma present. Koeppe gonioscopy is a useful technique for this purpose, in both the office and operating room (Figs. 12.11 and 12.12). The iris and angle structures should be inspected carefully and the results recorded. The RetCam® digital imaging system (Clarity Medical Systems, Pleasanton, CA) can also be used in the operating room to photograph angle structures for documentation and future comparison.
FIGURE 12.11. Operating room gonioscopy using Koeppe lenses and a portable slit lamp provides vital diagnostic information in preparation for glaucoma surgery.
FIGURE 12.12. Goniophotographs of eyes with congenital glaucoma. A: Appearance often seen in lightly pigmented races, showing scalloped border of ciliary body band, no visible scleral spur, and uniform membrane-line appearance of trabecular meshwork band. This eye belongs to the infant shown in Figure 12.1, after initial goniotomy surgery. The cornea is still hazy, and a cleft is visible in the anterior trabecular meshwork at the arrow. B: Appearance of angle in a darkly pigmented eye, showing ciliary body band with uveal processes and a narrow trabecular meshwork band, without distinct scleral spur in either A or B.
The most characteristic feature of the child’s anterior chamber angle is the trabecular meshwork, which has the appearance of a smooth, homogeneous membrane, extending from peripheral iris to Schwalbe line, during the first year of life. It becomes coarser and more pigmented with the passing years. In addition, the peripheral iris in the young child tends to be thinner and flatter (18).
The iris in infantile glaucoma often shows a more anterior insertion than that of the normal infant, with altered translucency of the angle face producing an indistinct ciliary body band, trabecular mesh, and scleral spur. This translucent tissue has historically been referred to as “Barkan membrane” (19). The angle may show other characteristics suggestive of the etiology of glaucoma. For example, in glaucoma after cataract surgery, a closed angle with iris bombe suggests pupillary block and the need for peripheral iridectomy/synechiolysis, while an open angle suggests trabecular meshwork dysfunction and the need for a different treatment strategy. An abnormally prominent Schwalbe line and iris adhesions to the angle structures may alternatively suggest Axenfeld-Rieger syndrome (also known as iridocorneal dysgenesis). Juvenile open-angle glaucoma (JOAG) patients usually demonstrate a normal-appearing open angle, often with a prominent, lacy uveal meshwork.
Taken together with other findings of anterior examination (above), the adequacy of the angle view and its findings are important guides to the appropriate surgical intervention that may be needed.
Funduscopy usually concentrates on a careful assessment of the appearance of the optic nerve head. Other fundus features (e.g., a stalk in persistent fetal vasculature, choroidal hemangioma in Sturge-Weber syndrome) may help confirm the type of glaucoma or help with surgical planning. Large size of the optic nerve cup and asymmetry of cupping between fellow eyes is suggestive but not definite evidence of glaucoma. Illustratively, the cup/disc ratio exceeded 0.3 in 68% of 126 eyes with primary infantile glaucoma examined by Shaffer and Hetherington(20), but in only 2.6% of 936 normal newborn eyes examined by Richardson (21). Marked optic cup asymmetry was noted in only 0.6% of normal eyes in the latter series, contrasted with 89% noted for infants with monocular glaucoma. Indirect ophthalmoscopy using a 28 or 20 diopter condensing lens in the office often underestimates the optic nerve cupping, which more accurately can be assessed by slit-lamp biomicroscopy or with direct ophthalmoscopy through a Koeppe lens in the operating room (Fig. 12.13). In infants and young children, initial findings can be usefully compared with changes seen after control of glaucoma (or failure of control) as a measure of success of treatment; dramatic reversal of cupping can occur in young eyes after IOP reduction, likely due to an incompletely developed or very flexible lamina cribrosa (22). Drawings (and when possible, photographs) of the optic nerves are valuable for later comparison.
FIGURE 12.13. The appearance of the optic nerve can be well seen through a Koeppe lens. This helps neutralize the changes in size due to refractive error, and facilitates the exam through a fairly small pupil. Drawings of the cupping are helpful, as are photographs that allow future comparisons to be made.
Refraction, Perimetry, Ultrasound, Corneal Pachymetry, and Optical coherence tomography: Determination of refractive errors can be helpful, especially when they are asymmetric in the setting of probable unilateral glaucoma; in this case, relative myopia of the affected eye supports the diagnosis of glaucoma. Visual field testing (using Goldmann kinetic or automated techniques) can be useful in cooperative older children with known glaucoma, allowing assessment of the extent of initial field loss, as well as stability of the remaining visual field over time. Axial length measurement with ultrasound (during examination under anesthesia) can be an adjunct to serial corneal diameter measurements when following infants and young children being treated for glaucoma (17). Ultrasound pachymetry (to measure the central corneal thickness) has proven relevant to evaluation of IOP in cases of adult open-angle glaucoma, especially when the cornea is much thinner or thicker than average; thinner central corneas tend to produce an underestimation of the true IOP by applanation, but also seem to confer additional risk for glaucomatous damage as an independent risk factor in adults (23,24,25). Several studies have reported on central corneal thickness in normal children, as well as in those with glaucoma and ocular diseases associated with childhood glaucoma (26,27,28,29). Decreasing target IOP for children’s eyes with thinner than average central corneas may be advisable, but the measured IOP should not be “adjusted” downward for eyes with thick corneas, especially in the presence of conditions such as aphakia, where thicker corneas may be acquired rather than congenitally occurring (28).
Optical coherence tomography (OCT) (Carl-Zeiss Meditech, Dublin, CA), a non-contact imaging technology capable of measuring the thickness of the peripapillary retinal nerve fiber layer and the macular area in both adults and children (30,31,32), correlates with photographic and visual field evidence of glaucomatous optic nerve head damage(33) and may prove valuable to evaluate the thinning of these parameters in children with glaucoma (33,34).
Many conditions of the eye may produce symptoms and ocular signs that suggest possible glaucoma, including simple congenital nasolacrimal duct obstruction, corneal disease, or anterior chamber inflammation, which may variably produce photophobia and tearing. Storage diseases associated with corneal clouding or hereditary corneal dystrophies with opacification of the cornea raise concern for glaucoma in a child, as does isolated corneal enlargement. It is important always to rule out glaucoma when any of these signs or symptoms is present, even when initial evidence suggests a more common nonglaucomatous cause. Glaucoma may also complicate inflammation or trauma to the anterior segment, may be found with storage diseases and with primary enlargement of the cornea, and certainly is seen coincidentally with lacrimal duct obstruction (35).
In summary, the signs of glaucoma are shared by other eye diseases and indicate the careful anterior segment examination and tonometry necessary to rule out this abnormality. The identification of some other cause of such signs does not in itself eliminate the risk of glaucoma.
PRIMARY CHILDHOOD GLAUCOMA
Primary Congenital Open-Angle Glaucoma
This disorder is the most common primary pediatric glaucoma, with an estimated incidence of only about one in 10,000 to 20,000 live births in Western countries, but with a higher incidence in Middle East and Slovak Romany populations (1). There is no predilection for race or gender, and most cases (65% to 80%) are bilateral (3). More than 80% of all cases have an onset of disease within the first year of life, with about 25% diagnosed as newborns, and more that 60% presenting by 6 months of age (3,36). Primary congenital (also called primary infantile) glaucoma may present with severe phenotype at birth, or may present with milder signs and symptoms, leading to delay in its diagnosis, sometimes after significant and irreversible optic nerve damage has occurred from chronically elevated IOP. Typically, the corneal abnormalities consisting of progressive edema associated with breaks in Descemet membrane occur during the first year of life. Recognition of glaucoma depends on the sensitivity of caretakers to the significance of these signs and symptoms.
While the majority of primary infantile glaucoma cases are sporadic (no known family history), about 10% are familial, usually transmitted as an autosomal recessive trait, with penetrance varying from 40% to 100% (37,38). Several genetic loci associated with primary congenital glaucoma have been identified in large pedigrees using linkage analysis. Two main causative genes have thus far been reported, the CYP1B1 gene (a cytochrome P450 system enzyme) and the LTB2 gene (39); the MYOC gene has also been implicated in a minority of primary congenital glaucoma cases (40). Mutations in the CYP1B1 gene have been identified in many cases of primary congenital glaucoma worldwide (41,42,43,44), but do not seem responsible for many sporadic cases in the United States (unpublished personal data) (45).
Table 12.2 DIFFERENTIAL DIAGNOSIS OF FEATURES COMMONLY FOUND IN PEDIATRIC GLAUCOMAS
Disorders with “red eye” and/or epiphora and/or photophobia
The significant inherited defect is confined to the filtration tissues, rendering them less permeable to the passage of aqueous humor. The gonioscopic abnormality typically features decreased transparency of the tissues over the scleral spur and ciliary body band, so that these normal angle landmarks are difficult to define. The width of the trabecular meshwork and ciliary body also may be diminished, giving the impression of an anterior insertion of the iris (see above under gonioscopy).
Surgical intervention is the definitive treatment for primary congenital glaucoma, with angle surgery (usually goniotomy or trabeculotomy) successful in the majority of cases, especially those with presentation between 3 and 12 months of age; surgical success drops for those with presentation at birth or after age 1 to 2 years (see below). In cases refractory to angle surgery, filtration surgery (sometimes combined with trabeculotomy) (46,47,48,49,50), glaucoma implant surgery (49,51,54,55,56,57), and cycloablation (58) have been used with variable success, depending upon the reported series (see later under Treatment). Visual prognosis is dependent not only upon the timely diagnosis and IOP reduction, but upon the secondary corneal, refractive, and optic nerve changes produced by the initially elevated IOP (3).
Juvenile Open-Angle Glaucoma
In contrast to primary congenital or infantile glaucoma, this rare disease is an autosomal dominant early onset form of primary open-angle glaucoma. JOAG is characterized by acquired and marked bilateral IOP elevation, with usual onset between ages 4 and 35 years, often with a strong family history. Ocular damage, usually in the form of optic nerve cupping and visual field loss, is usually asymptomatic, unless myopia brings the child to eye examination for decreased distance vision. Absent are the corneal stigmata usually present in infant-onset glaucoma. Gonioscopy reveals normal-appearing angle structures. Treatment is difficult, often beginning with medication, and proceeding to filtration or tube implant surgery, although angle surgery may be helpful in some cases (see below).
JOAG was first linked to chromosome 1q21-31 by Sheffield et al. in 1993 (59). Mutations were subsequently reported in the trabecular meshwork glucocorticoid response gene (TIGR, now renamed myocilin) and have been identified in numerous populations of individuals with JOAG worldwide (60,61,62,63).
Primary Pediatric Glaucoma Associated with Systemic Diseases
Primary glaucoma in children may be seen in association with certain systemic diseases in which ocular abnormalities are included in the syndrome complex (see Table 12.1).
Sturge-Weber Syndrome
Glaucoma is present commonly with this disease in association with a facial nevus flammeus of the ipsilateral upper eyelid, and abnormal vasculature of the leptomeninges. Intracranial involvement may be complicated by epilepsy, paralysis, and visual field defects. The glaucoma may be congenital or acquired and is most often unilateral. The onset of glaucoma seems bimodal, with some cases presenting in early infancy, while others occur later in childhood. Inspection of the conjunctiva often shows an abnormal number and tortuosity of blood vessels with elevation of venous pressure. A striking episcleral vascular abnormality is present behind the limbus circumferentially. Gonioscopy reveals minor angle anatomic changes, usually without vascular abnormalities of the angle; blood can often be identified in Schlemm canal. The iris of the involved eye is often more pigmented than that of the fellow eye. Funduscopy generally reveals evidence of a choroidal hemangioma and disc changes secondary to glaucoma. The etiology of glaucoma in Sturge-Weber syndrome is most often considered to be increased episcleral venous pressure secondary to the ipsilateral choroidal hemangioma, although congenitally abnormal angle structures may contribute to infant-onset disease (64,65).
Only gold members can continue reading. Log In or Register to continue