7 ![]()
Two conditions with disorders of the corneal endothelium are discussed in this chapter. Both iridocorneal endothelial (ICE) syndrome and posterior polymorphous dystrophy are conditions in which there is strong clinical and histopatholigic evidence that the corneal endothelial disorder is directly associated with the changes causing the glaucoma.
Definition
The ICE syndrome includes several related disorders: essential iris atrophy or progressive iris atrophy, iris nevus (Cogan-Reese) syndrome, and Chandler’s syndrome. Glaucoma is an important feature of this syndrome, and the related disorders may represent a continuum of the same process. Scheie and Yanoff1 and Yanoff2 proposed the term ICE to group essential iris atrophy, Chandler’s syndrome, and iris nevus syndrome as varying clinical manifestations of a single disease entity.
What Is Progressive (Essential) Iris Atrophy?
A form of secondary glaucoma associated with atrophy of the iris and hole formation was described by Harms3 in 1903. The condition was termed essential iris atrophy or progressive essential iris atrophy. Progressive iris atrophy is characterized by prominent atrophy of the iris, with iris stromal and/or full-thickness iris holes. Peripheral anterior synechiae develop early, and progress both circumferentially and onto the cornea.4 Pupillary distortion and ectropion uveae occur typically toward the most prominent anterior synechiae.4,5 More specifically, progressive iris atrophy leads to two types of holes: stretch holes and melting holes. Stretch holes are formed in the iris from tractional forces between peripheral anterior synechiae on opposite sides of the anterior chamber. Melting holes may also develop in the vicinity of peripheral anterior synechiae (Fig. 7–1).
What Is the Iris Nevus (Cogan-Reese) Syndrome?
In 1969, Cogan and Reese6 described two patients with pigmented nodules of the iris, associated with some features similar to essential iris atrophy and Chandler’s syndrome. In both cases, melanoma was suspected and the eyes were enucleated. However, on histopathologic examination, the nodules were composed of benign tissue resembling iris stroma. Later studies revealed that these nodules could occur on the iris surface in association with the spectrum of changes seen with essential iris atrophy and Chandler’s syndrome. As such, the condition became known as Cogan-Reese syndrome. Scheie et al7 described similar cases with diffuse iris nevi instead of nodules on the surface of the iris. They called this condition the iris nevus syndrome.
Iris pigmented lesions ranging from multiple, pedunculated, nodular lesions to diffuse, smooth, velvety changes are present. The iris surface often loses its normal architecture and appears darker than the iris of the fellow eye. Ectropion uveae, breaks in the iris stroma, and an ectopic pupil are often present. Also, peripheral anterior synechiae, corneal edema, and glaucoma are characteristic features (Fig. 7–2).
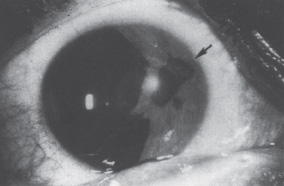
Figure 7–1. Large peripheral anterior synechiae (arrow) with correctopia, iris holes, and ectropion uveae in a patient with essential iris atrophy. (With permission from Miller CA, Krachmer JH: Endothelial dystrophies. In: Kaufman HE, Barron BA, McDonald MB (eds): The Cornea, 2d Ed. Newton, MA: Butterworth-Heinemann, 1997;470.)
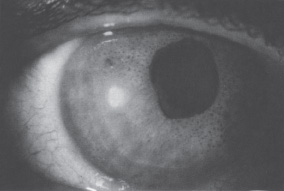
Figure 7–2. Numerous dark brown iris nodules, ectropion uveae, and correctopia in a patient with iris nevus syndrome. (With permission from Miller CA, Krachmer JH: Endothelial dystrophies. In: Kaufman HE, Barron CA, McDonald MB (eds): The Cornea, 2d Ed. Newton, MA: Butterworth-Heinemarm, 1997;471.)
What Is Chandler’s Syndrome?
In 1956, Chandler8 reported cases that were similar to essential iris atrophy but different in that changes in the iris were limited to slight correctopia and mild stromal atrophy. Here, he noticed that corneal edema appeared to be a more consistent feature and often occurred at intraocular pressures that were normal or only slightly elevated. In all cases, Chandler noted an abnormality of the corneal endothelium, which was described as having the appearance of fine, hammered silver. The condition became known as Chandler’s syndrome. In both essential iris atrophy and Chandler’s syndrome, glaucoma is associated with progressive closure of the anterior chamber angle. Iris atrophy is less prominent in Chandler’s syndrome than in progressive iris atrophy and, when detectable, is often limited to the anterior iris stroma.9,10 The pupil is usually round or slightly oval.
What Are the Essential Features of the ICE Syndrome?
The ICE syndrome is characterized by the proliferation and spreading of an abnormal corneal endothelial membrane across the iridocorneal angle and iris surface. This “endothelialization” often results in iridocorneal adhesions, glaucoma, pupillary distortion, iris atrophy and corneal decompensation. The primary corneal abnormality is usually unilateral, nonfamilial, and often rapidly progressive. However, cases of bilateral involvement have been described.11 Although development of peripheral anterior synechiae is common, the accompanying secondary glaucoma is frequently more advanced than would be expected by the extent of the synechiae, presumably because of the angle endothelialization11 (Fig. 7–3). Common manifestations of ICE syndrome also include, in addition to abnormalities of the iris, reduced visual acuity and pain.
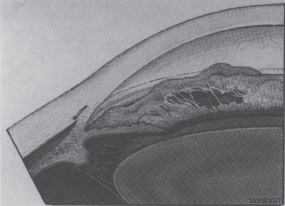
Figure 7–3. The angle in essential iris atrophy. (With permission from Kanski JJ: Clinical Ophthalmology, 2d Ed. London: Butterworth, 1989;222.)
Epidemiology and Importance
What Is the Epidemiology of the ICE Syndrome?
Classically young to middle-aged women are affected, although men can also be affected. The male/female ratio varies from 1:2 to l:5.12 The typical patient is a white woman with unilateral disease and a negative family history. The onset of symptoms occurs in early to middle adulthood. Unlike posterior polymorphous dystrophy, there does not appear to be a genetic predisposition for iridocorneal endothelial syndrome. In posterior polymorphous dystrophy, a small percentage of cases have been linked to Alport’s syndrome.13 Linkage of posterior polymorphous to the long arm of chromosome 20 (20q11) was reported in a family who had 21 members with this disorder.14
What Were Previous Theories of Pathogenesis Regarding the ICE Syndrome?
The etiology of the ICE syndrome has been controversial since the condition was first described. Previous popular theories are enumerated in Table 7–1. It was originally thought that iridocorneal endothelial syndrome was the result of low-grade intraocular inflammation.11 Most clinical cases reviewed did not show evidence of active inflammation, and the histologic studies have not supported a primary inflammatory cause.2,7,15–17 Nevertheless, in true cases of essential iris atrophy, it is possible that ciliary flush may be associated with advanced bullous keratopathy. Also low-grade, chronic aqueous flare may be associated with the leaking of iris vessels; therefore, occasional evidence of intraocular inflammation does not rule out the diagnosis of the ICE syndrome.2
Vascular insufficiency of iris vessels has also been postulated as a mechanism of essential iris atrophy. Ischemia from sclerosis of iris vessels, congenital vascular disturbances, and vascular toxins have been proposed as theories. Anterior segment fluorescein angiography demonstrated vascular abnormalities of the iris in essential iris atrophy.17 These abnormalities suggest the possibility of secondary alterations of the iris vasculature.
The membrane theory of Campbell17a was based on a clinical study of 82 patients with essential iris atrophy and a histopathologic study of 10 enucleated eyes with this condition. He postulated that essential iris atrophy begins as an abnormality of the corneal endothelium, which leads to corneal edema. The membrane sometimes extends across the angle and over the iris surface. When the membrane contracts, peripheral anterior synechiae develop and pupillary distortion occurs. The iris in the opposite quadrant undergoes thinning with variable degrees of atrophy. The aforementioned process can lead to irregular holes in the iris.
What Is the Current Theory Regarding the Etiology of the ICE Syndrome?
Leibowitz and Waring18 have proposed that an acute pathologic insult to the corneal endothelium occurring in the second or third decade of life, for example, a viral endotheliitis, may damage the endothelium and convert it to ICE cells. These ICE cells exhibit migratory behavior and elaborate aberrant basement membrane and collagenous tissue. ICE cell alterations occur in corneas with antecedent herpes simplex keratouveitis and glaucoma.19 Using the polymerase chain reaction to identify viral DNA in corneal specimens from patients with the iridocorneal endothelial syndrome, 16 of 25 iridocorneal endothelial syndrome patients and four of six patients with herpetic keratitis specimens were positive for herpes simplex virus.19 When the endothelium was removed, the positive specimens became negative, thus localizing herpes simplex DNA to the endothelium. These data, although extremely compelling, do not provide definitive evidence that herpes simplex causes iridocorneal endothelial syndrome. Instead there are three possible interpretations of the findings19 (Table 7–2).
The hypothesis that herpes simplex virus may cause the iridocorneal endothelial syndrome is consistent with the acute onset, mild inflammation, and unilaterality of this disorder. Epstein-Barr virus has also been implicated in possibly causing iridocorneal endothelial syndrome.20 Once the endothelial cells are damaged, they exhibit migratory and secretory behavior and produce clinical iridocorneal endothelial syndrome. The ICE cells are of variable shapes, sizes, and density. There is disagreement about whether the cells are actually epithelial-like; however, there appears to be little evidence that these cells are of direct epithelial origin.
| Inflammatory theory |
| Vascular theory |
| Primary iris defect theory |
| Membrane theory of Campbell |
| Herpes simplex is one of the initiators of the ICE syndrome |
| The process producing the ICE syndrome activates latent herpes simplex virus and contributes to its pathogenesis |
| The processes producing the ICE syndrome activate latent herpes, but the virus is not involved in the pathogenesis of the ICE syndrome |
What Is the Value of Specular Microscopy in the ICE Syndrome?
Specular microscopy contributed greatly to understanding and diagnosing the ICE syndrome.21 As a noninvasive, painless, outpatient technique, it is used to obtain high-resolution microscopic images of the corneal endothelium. Specular microscopy performed on patients with ICE syndrome demonstrates a population of abnormal cells called ICE cells.22 ICE cells, which are pathognomonic of ICE syndrome, are larger and more pleomorphic than normal corneal endothelial cells. These cells are observed in most cases, although they are not readily discernible in patients with severe corneal edema. In some patients, the normal endothelial architecture is replaced completely by ICE cells (total ICE), whereas in others, normal endothelial cells are only partially replaced (subtotal ICE)22,23 (Fig. 7–4).
What Is Posterior Polymorphous Dystrophy?
Deep corneal lesions of various shapes characterize posterior polymorphous dystrophy (PPD). Nodular, grouped vesicular and blister-like lesions commonly occur.24 Gray-white halos often surround the vesicular lesions. Flat gray-white opacities, gray thickenings of Descemet’s membrane, sinuous broad bands, or clear bands with white scalloped margins also can be present.25 These bands can be oriented vertically or horizontally and often are confused with Descemet’s tears.26 The lesions are easily seen on retroillumination. On specular microscopic evaluation, the lesions contain abnormal, pleomorphic cells, with indistinct borders and increased reflective highlights.
This dystrophy is usually transmitted as a dominant trait, but recessive patterns of inheritance have been reported.26 It is usually bilateral, but can be asymmetric and, rarely, unilateral.23 Congenital cases do exist and present with cloudy corneas at birth. Most patients with PPD have normal vision and are asymptomatic, making the age of onset difficult to determine. Most cases are nonprogressive, but in some cases endothelial decompensation can develop.25
PPD can be associated with anterior segment dysgenesis, with prominent Schwalbe’s ring, iridocorneal adhesions, abnormal iris processes, iris atrophy, correctopia, and ectropion uveae.25 Intraocular pressure (IOP) elevation occurs in approximately 15% of cases.25 The pathogenesis of PPD is unknown, but may involve endothelial cells undergoing transformation into epithelial-like cells. The angle and iris abnormalities may represent spread of these abnormal cells from the cornea.
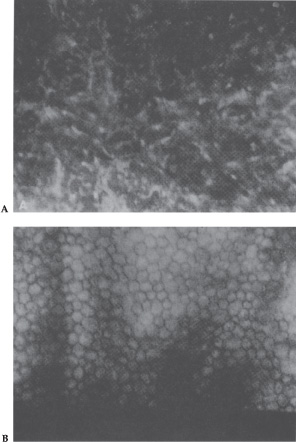
Figure 7–4. (A) Specular microscopy of the corneal endothelium in the ICE syndrome. Cell borders are obscured, resulting in loss of the normal endothelial mosaic. Note dark areas within endothelial cells. Brighter reflections are believed to be from cell borders. (B) Specular microscopy of fellow eye showing normal endothelial mosaic. (With permission from Mandelbaum S: Glaucoma associated with corneal disorders. In: Duane’s Clinical Ophthalmology, Vol. 3. Philadelphia: JB Lippincott, 1993; Ch. 54F:3.)
Diagnosis and Differential Diagnosis
How Do the ICE Syndrome and PPD Differ?
The majority of ICE and PPD cases can be distinguished clinically on slit-lamp examination. PPD is a bilateral familial disorder (Table 7–3). It has been diagnosed in all age groups, usually as an incidental finding, because it is typically asymptomatic. Commonly nonprogressive, only occasionally is it associated with corneal decompensation or glaucoma. The posterior corneal abnormality shows clusters of vesicles and occasional excresences of Descemet’s membrane.8
The universal clinical sign of the iridocorneal endothelial syndrome is a finely hammered silver appearance of all or part of the posterior corneal surface when viewed in specularly reflected light with the slit lamp.27,28 ICE cells give rise to a hammered silver appearance on slit-lamp exam. They appear as a negative of normal endothelial cells on specular microscopy and are pathognomonic of the ICE syndrome. Classically, the diagnosis of PPD requires the presence of small, round, discrete, transparent vesicular lesions, surrounded by a ring of opacity at the level of Descemet’s membrane. In some cases, however, confusion and misdiagnosis occur. Corneal edema may obscure posterior corneal details. Peripheral anterior synechiae, ectropion uveae, and secondary glaucoma may be present creating a clinical picture similar to that of iridocorneal endothelial syndrome (Table 7–4).
What Are the Associations Between Glaucoma and the Iridocorneal Endothelial Syndrome?
Few previous studies have focused on the issue of glaucoma secondary to the ICE syndrome from a clinical standpoint. Case reports indicate that glaucoma associated with the ICE syndrome is difficult to control and can result in marked visual impairment.29–31 Therefore, it is of paramount importance to identify those patients who are most likely to develop glaucoma and devise treatment plans accordingly. The reported prevalence of glaucoma associated with the ICE syndrome is 37 to 82%.31
Feature | ICE | PPD |
Laterality | Unilateral | Bilateral |
Symptom onset | 2d to 3d decade | Any age |
Heredity | None | Autosomal dominant |
Sex | Females > males | Female = male |
Manifestation | ICE | PPD |
Corneal edema | Common | Occasional |
Endothelium | Guttate-like changes | Ridges, vesicles, plaques |
Iridocorneal adhesions | Common | Occasional |
Iris stromal atrophy | Mild to marked | Minimal or absent |
Ectropion uveae | Common | Rare |
Glaucoma | Very common | Rare |
The diagnosis of iridocorneal endothelial syndrome should be considered in younger patients with unilateral glaucoma and confirmed by specular microscopy (Table 7–5). Glaucoma results from obstruction of the anterior chamber angle by an abnormal membrane and often closure of the angle by peripheral anterior synechiae.
One must consider posterior corneal abnormalities in the differential diagnosis of ICE. These include Fuchs’ corneal endothelial dystrophy, PPD, congenital glaucoma, congenital hereditary endothelial dystrophy, trauma, and postsurgical abnormalities. Fuchs’ dystrophy is a bilateral, often asymmetric condition. It can be associated with hypermetropia, short axial length, and shallow anterior chamber.32 Women are affected more severely and 2½ times more frequently than men.33 The earliest finding of corneal guttae is not specific for this disease. Most patients with guttae never develop the classic signs of Fuchs’ dystrophy. In some patients, the guttae lead to corneal endothelial cell loss and an inability to maintain corneal clarity. When this happens, stromal and epithelial edema usually result and can often accompany corneal scarring.
Posterior corneal abnormalities |
Fuchs’ endothelial dystrophy |
Posterior polymorphous dystrophy |
Congenital (CHED, congenital glaucoma) |
Traumatic (blunt, chemical, radiation) |
Postsurgical |
Iris atrophy |
Rieger’s syndrome |
Aniridia |
Herpetic ocular infections |
Iridocyclitis |
Xeroderma pigmentosa |
Iridoschisis |
Congenital hypoplasia of the iris stroma |
Iris nodules |
Melanomas |
Iris nevi |
Neurofibromatosis |
Inflammatory nodules (Lisch) |
Flocculus of the iris |
Juvenile xanthogranuloma |
CHED, congenital hereditary endothelial dystrophy. |
PPD is a bilateral, dominantly inherited disorder of the corneal endothelium, characterized by a spectrum of changes on the posterior corneal surface, and less commonly in the iris and anterior chamber angle.24 Sometimes changes in the fellow eye are so minimal that it can be diagnosed as unilateral. PPD usually does not progress to corneal edema and most cases are asymptomatic. Congenital hereditary endothelial dystrophy is the most commonly encountered congenital dystrophy. It occurs in both dominant and recessive forms. The dominant form is more severe and usually present at birth.34 The entire corneal stroma is markedly edematous and the cornea takes on a milky appearance. Throughout life, there is little variation in the clinical appearance. A spectrum of severity varying from minimal to severe can be observed in families with the dominant form of the disorder, and the parents often have PPD changes in the cornea.35 Any form of trauma can damage the endothelium. Blunt trauma can cause ruptures of Descemet’s membrane with the subsequent formation of corneal edema. During surgery, Descemet’s membrane can become disinserted from the overlying stroma which can lead to corneal edema.
Iris atrophy is also included in the differential diagnosis of ICE. Essential or progressive iris atrophy is often seen with the ICE syndrome; however, it can also been seen in other forms of ocular pathology. Rieger’s syndrome, aniridia, herpes, iridocyclitis, xeroderma pigmentosa, iridoschisis, and congenital hypoplasia of the iris stroma must all be considered in the differential diagnosis of essential or progressive iris atrophy. The three cardinal features of Rieger’s anomaly are posterior embryotoxon with iris adhesions, hypoplasia of the iris stroma, and bilateral involvement. Ocular-associated findings include dyscoria, glaucoma, and congenital anterior synechiae. Systemic associations include Marfan syndrome, Ehlers-Danlos syndrome, Down syndrome, and oculodentodigital dysplasia.36 Less frequently associated ocular findings include megalocornea, microcornea, aniridia, and Peters’ anomaly. Herpetic keratouveitis/iridocyclitis can cause iris atrophy. Herpes simplex virus can produce uveitis, although severe herpetic uveitis is rare. In some patients, the iris may develop segmental necrosis, whereas in others, the entire iris may develop intravascular thromboses and become ischemic and necrotic.
Xeroderma pigmentosa is an autosomal recessive disorder caused by a defect in the ability to repair DNA damaged by ultraviolet light. Ocular findings include eyelid tumors, especially basal and squamous cell carcinomas. Anterior segment involvement includes conjunctival inflammation and membrane development, corneal scarring and vascularization, and anterior segment inflammation, which can subsequently lead to iris atrophy. Treatment includes prevention of tumors by the use of sunscreening agents and avoiding exposure to sunlight. Penetrating keratoplasty for corneal scarring has a poor prognosis.37 Iridoschisis and congenital hypoplasia of the iris stroma must also be considered in the differential of iris atrophy.
Iris lesions that vary in number, size, shape, location, and texture typify the iris nevus syndrome of Cogan-Reese. Included in the differential dianosis of iris nodules are the following: iris nevi, melanomas, neurofibromatosis, inflammatory nodules (Lisch), and juvenile xanthogranuloma. Small malignant melanomas of the iris may be impossible to differentiate clinically from benign iris nevi and other simulating lesions. Signs suggestive of malignancy include extensive ectropion iridis, prominent vascularity, sector cataract, secondary glaucoma, documented progressive growth, and lesion size. Iris melanomas range in appearance from amelanotic to dark-brown lesions. In rare instances, they assume a diffuse growth pattern, producing a syndrome of unilateral acquired hyperchromic heterochromia and secondary glaucoma. An iridectomy needs to be performed for any lesion in which there is suspected growth. If the tumor extends into the anterior chamber angle, an iridotrabeculectomy or iridocyclectomy can be performed. Enucleation appears to be indicated for eyes containing a diffuse iris melanoma, especially in glaucomatous eyes. The prognosis for most patients with iris melanomas is excellent because the biologic behavior of most of these iris tumors is usually benign. Iris nevi are discrete masses or nodules on the anterior surface of the iris. These lesions tend to be variably pigmented and composed of benign nevus cells. There is an increased incidence of iris nevi in patients with neurofibromatosis. In neurofibromatosis, one can see multiple lesions varying from tan to dark brown and about the size of the head of a pin. These lesions may be flat or elevated and are referred to as Lisch nodules.
Juvenile xanthogranuloma typically affects the uveal tract, but may also affect the conjunctiva and the corneal stroma. Yellowish-to-gray, poorly demarcated iris lesions associated with raised organ skin lesions appear within the first year of life. The lesions may be associated with spontaneous hyphema and secondary glaucoma. Histopathologically, there is a diffuse granulomatous infiltrate with lipid containing histiocytes and Touton giant cells. These lesions regress spontaneously and may also be found in the ciliary body, anterior choroid, episclera, cornea, lids, and orbit.
Treatment and Management
How Is Glaucoma Associated with the ICE Syndrome Treated?
Glaucoma in the ICE syndrome results from progressive loss of the anterior chamber angle. Formation of peripheral anterior synechiae increases with time. IOP may be higher than expected based on the area affected by the synechiae. Histologic study has confirmed that the abnormal endothelium and basement membrane often overlie the trabecular meshwork even when the angle appears open on gonioscopy.28,31 It is postulated that this membrane consisting of ICE cells interferes with aqueous outflow even prior to the development of synechiae and contributes to elevated IOP (Fig. 7–5).
In the early stages, glaucoma may be controlled medically. Drugs decreasing aqueous production are more useful than miotics because the angle is often closed by synechiae or covered by an abnormal membrane. However, medical management has been found generally ineffective over the long term.30,31 Argon laser trabeculoplasty is generally not effective and should be avoided. Therefore, if the IOP cannot be controlled medically, filtering surgery is indicated. Controversy exists in the reports of success rates in the ICE syndrome. One study reports initial success rates for trabeculectomy in the ICE syndrome as comparable to that for primary open-angle glaucoma.38 However, others contend success rates are very poor for trabeculectomy in the ICE syndrome.38 Late failures are usually attributed to proliferation of abnormal endothelium into the filtering bleb.31 Progressive endothelialization of the blebs and the young age of the patients with the ICE syndrome are also common explanations for bleb failure. In these cases, another filtering procedure performed in a different location is appropriate. The success rates for repeated trabeculectomies are comparable to those of the initial procedure.38 The use of antimetabolites and setons may improve the results of secondary filtering procedures in recalcitrant cases.39 Vigorous control of inflammation following filtering procedures is necessary to help avoid bleb failure. Because the viral etiology of this syndrome has yet to be definitively confirmed, topical and systemic antiviral treatment may be therapeutic.40,41
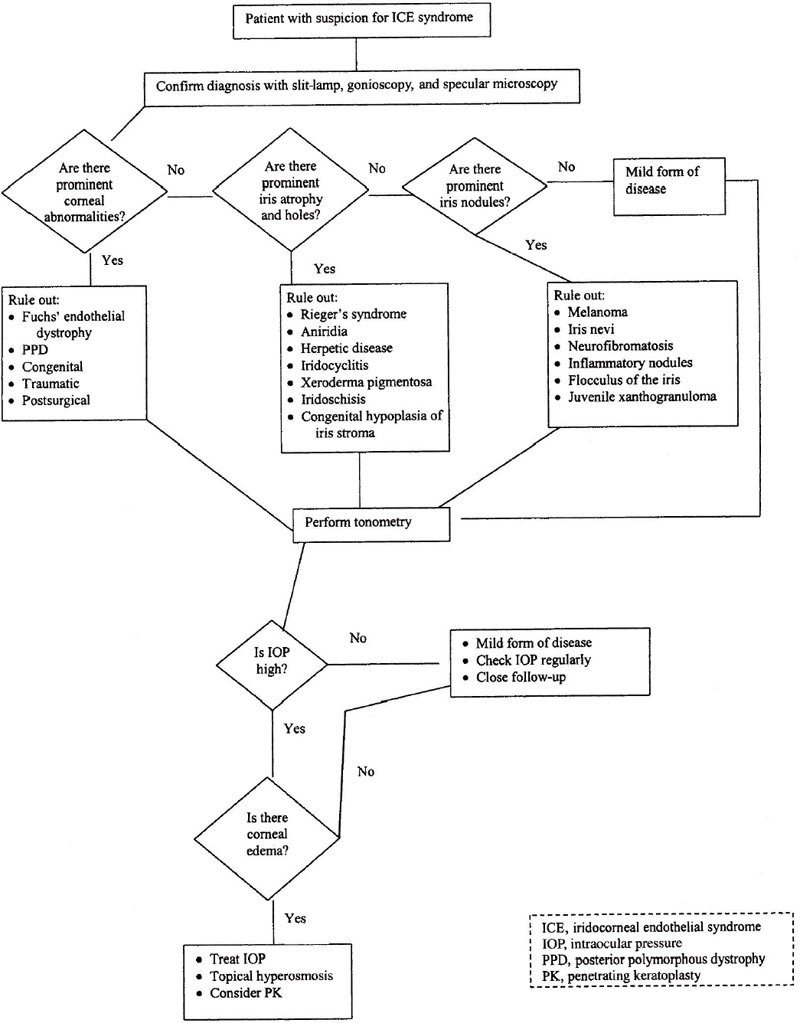
Figure 7–5. Management of a patient with the ICE syndrome.
Patients with corneal edema may benefit from lowering of the IOP. However, filtering surgery cannot be recommended solely in an attempt to resolve the corneal edema.42 The cornea can remain edematous even at the lowest achievable levels of IOP. Hypertonic saline drops are often helpful for mild corneal edema. If visually significant corneal edema is present after IOP has been lowered medically, then penetrating keratoplasty is usually required (Fig. 7–6). If the IOP remains controlled, the prognosis for the corneal graft is good.43 Recurrences of the endothelial abnormalities that characterize the ICE syndrome have not been noted to develop on the donor cornea.27,43
What Is the Role of Penetrating Keratoplasty in the Management of the ICE Syndrome?
Corneal edema, even at normal intraocular pressures, predominates in Chandler’s syndrome. Early corneal edema may respond to topical hyperosmotic solutions or ointment. Lowering the IOP improves corneal clarity and visual acuity in some patients. Penetrating keratoplasty is indicated in cases in which corneal edema significantly reduces vision, precludes visualization of the optic nerve, or causes pain from bullous keratopathy, recurrent corneal erosions, or secondary infectious keratitis.43–45 The success rate for penetrating keratoplasty in the ICE syndrome has generally been favorable despite poor overall clinical prognosis for the disorder.43–45 The role of surgical pupilloplasty and lysis of peripheral anterior synechiae at the time of penetrating keratoplasty remains unclear.
What Is the Summary of Treatment of the ICE Syndrome?
The most important problem to treat in the ICE syndrome is elevated IOP. Treatment is more difficult than that of chronic open-angle glaucoma because an abnormal membrane and peripheral anterior synechiae cover the trabecular meshwork. If the IOP is controlled, corneal edema is often less severe. However, corneal edema can be visually significant and warrant penetrating keratoplasty in a small percentage of cases. Penetrating keratoplasty is usually successful in the variants with predominantly corneal disease; however, it may be less successful in those cases with extensive iris disease and glaucoma. Generally, we do not treat iris atrophy or ectopic pupils unless the pupil is significantly eccentric to interfere with visual acuity. In these cases, surgical or yttrium-aluminum-garnet (YAG) laser iridoplasty may be of value.
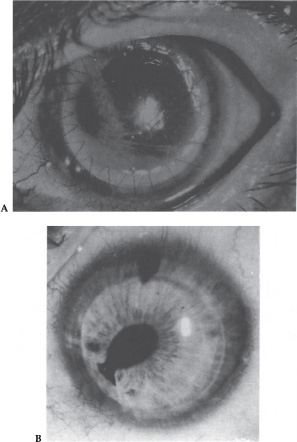
Figure 7–6. (A) Clear right corneal graft 1 month after penetrating keratoplasty for decompensated cornea due to iridocorneal endothelial syndrome. Marked iris dissolution and hole formation is apparent. (B) Left corneal graft for iridocorneal endothelial syndrome remains clear for 9 years after penetrating keratoplasty. (With permission from Crawford GJ, Stulting RD, Cavanagh HD, et al: Penetrating keratoplasty in the management of iridocorneal endothelial syndrome. Cornea 1989;8:34–38.)
1. Primary corneal endothelial disorders
|
2. Anterior segment developmental abnormalities
|
3. Congenital
|
4. Corneal and/or intraocular infectious/inflammatorv processes
|
5. Penetrating keratoplasty performed on eyes developing glaucoma |
Future Considerations
Future considerations include morphometric characteristics of the contralateral corneal endothelium in the ICE syndrome. Luca-Glass et al37 have tried to determine whether subclinical morphologic abnormalities exist in the contralateral, clinically uninvolved eye of patients with the ICE syndrome. Their data suggest the corneal endothelium of the clinically uninvolved, contralateral eye may have increase pleomorphism, expressed as a relatively low percentage of hexagonal cells. They found that this issue is important when considering the various hypotheses proposed to explain the pathogenesis of the disease. More studies are needed to further unravel this mystery.
References
1. Scheie HG, Yanoff M: Iris nevus (Cogan-Reese) syndrome. Arch Ophthalmol 1975;93:963–970.
4. Shields MB: The essential iris atrophies. Am J Ophthalmol 1978;85:749–755.
8. Chandler PA: Atrophy of the stroma of the iris, endothelial dystrophy, corneal edema and glaucoma. Am J Ophthalmol 1956;41:607–615.
16.Hetherington J: The spectrum of Chandler’s syndrome. Ophthalmology 1978;85:240–244.
23. Snell AC, Irwin ES: Hereditary deep dystrophy of the cornea. Am J Ophthalmol 1958;45:636–674.
29. Patel A, Kenyon KR, Hirst LW, et al: Chandler’s syndrome. Surv Ophthalmol 1983;27:327–344.
34. Maumenee AE: Congenital Hereditary corneal dystrophy. Am J Ophthalmol 1960;50:1114–1127.
39. Wright MM, Grajewski AL, Cristol SM, et al: 5-FU after trabeculectomy and the iridocorneal endothelial syndrome. Ophthalmology 1991;98:314–319.
45. Gaasterland DE, Rodrigues MM, Moshell AN: Ocular involvement in xeroderma pigmentosa. Ophthalmology 1982;89:980–987.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


