8 Glaucoma
INTRODUCTION
Visual loss in glaucoma is caused by the death of the retinal ganglion cells and their axons that constitute the nerve fiber layer of the retina and the optic nerve (Fig. 8-1A). The optic nerve head has a characteristic cupped or excavated configuration in glaucoma (Figs. 8-1B, 8-2, and 8-3B). Cupping distinguishes glaucomatous optic atrophy from primary optic atrophy, in which loss of retinal ganglion cells and nerve fibers also occurs. Cupping of the optic disc suggests that elevated intraocular pressure is a major risk factor in the pathogenesis of glaucomatous optic atrophy.
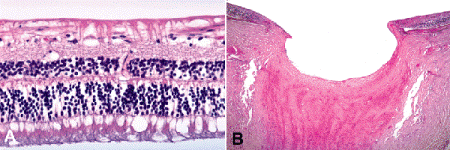
Fig. 8-1. A. Glaucomatous retinal atrophy. The ganglion cell and nerve fiber layers of the retina are atrophic. The inner plexiform and inner nuclear layers are well preserved excluding inner ischemic retinal atrophy. B. Glaucomatous optic atrophy. The nerve head is massively cupped, and the lamina cribrosa is bowed posteriorly. The nerve fiber layer of the retina is markedly atrophic. (A. H&E ×10, B. H&E ×100)
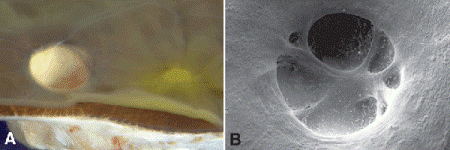
Fig. 8-2. A. Glaucomatous optic atrophy. The disc is pale and deeply cupped. Yellow macular pigment is seen temporally. B. Scanning electron microscopy of deeply cupped optic nerve.
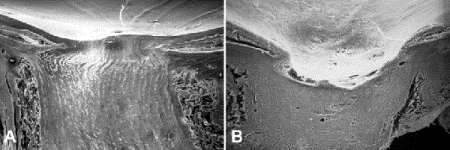
Fig. 8-3. Glaucomatous optic atrophy, SEM. A. Longitudinally sectioned normal nerve shows mild physiologic cupping. Lamina cribrosa is visible. B. Longitudinal section of nerve with severe glaucomatous cupping. Retina at margin of cup is severely atrophic.
How elevated intraocular pressure kills retinal ganglion cells is not clear. Experimental evidence suggests that it may be related to ischemia and/or blockage of axoplasmic flow caused by mechanical compression of axons in the pores of the lamina cribrosa, which are distorted by high levels of intraocular pressure. The blockage of axoplasmic flow may deprive cells of brain-derived neurotrophic factor whose absence triggers programmed cell death. Glial cell activation, TNF-α, and neuroinflammatory processes are thought to be important mediators of retinal ganglion cell damage.
Glaucoma has been defined as a syndrome characterized by an elevation of intraocular pressure of sufficient degree or chronicity to produce ocular tissue damage (Yanoff ) or as an optic neuropathy associated with a characteristic excavation of the optic disc and a progressive loss of visual field sensitivity (Quigley). The first definition emphasizes that tissue damage, usually nerve fiber atrophy or optic nerve cupping is a requisite for the diagnosis. The term syndrome indicates that there are many mechanisms that can raise intraocular pressure. The first definition also implies that a single elevated pressure reading is not glaucoma. The second newer definition does not mention intraocular pressure because authorities recently have stressed that elevation of intraocular pressure is only one of the risk factors that are responsible for neuronal loss in glaucoma. The latter definition includes so-called low-tension glaucoma that develops in patients whose optic nerves seem to be especially vulnerable to damage. Most glaucomatous eyes examined in the ophthalmic pathology laboratory have had elevated intraocular pressure. In nearly all cases of glaucoma, the elevated intraocular pressure is caused by obstruction of aqueous outflow.
As noted earlier (see Chapter 1), intraocular pressure is governed by a delicate balance between the production of aqueous humor by the nonpigmented ciliary epithelial cells and its egress or outflow from the eye via the trabecular meshwork and the canal of Schlemm, which are located in the anterior chamber angle formed by the cornea and peripheral iris (Figs. 1-9 and 8-4). Lesser amounts of aqueous exit through nontraditional pathways that include iris vessels and posterior uveoscleral outflow via the ciliary body and the vortex veins.
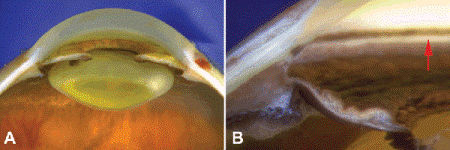
Fig. 8-4. A. Anterior segment. The anterior chamber is deep and the angle is open. The pupil is widely dilated. B. Anterior chamber angle. Trabecular meshwork is pigmented band directly in front of lighter scleral spur, which is marked by arrow. The meshwork is heavily pigmented in this eye.
The trabecular meshwork is a sieve-like structure that is nestled in the anterior crotch of the scleral spur (Fig. 1-9C,1-9D). It is composed of an interconnected network of small collagenous beams or trabeculae enveloped by trabecular endothelial cells. A thin layer of extracellular matrix material called the juxtacanalicular connective tissue (JXT) is interposed between the interstices of the meshwork and the lumen of Schlemm canal, which is lined by a continuous layer of endothelial cells (Fig. 8-5A). A modified vein, Schlemm canal runs circumferentially around the chamber angle, giving off branches or collector channels that traverse the sclera and discharge their contents into the epibulbar veins via the aqueous veins of Ascher.
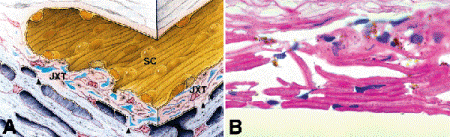
Fig. 8-5. A. Schlemm canal and JXT. Aqueous outflow obstruction in primary open angle glaucoma probably resides in the JXT, which borders the inner wall of Schlemm canal. Alvarado has shown that the area of the trabecular culs-de-sac is markedly reduced in primary open-angle glaucoma. These trabecular culs-de-sac, which abut the JXT, are responsible for a major proportion of normal outflow resistance. (From Alvarado JA, Murphy CG. Outflow obstruction in pigmentary and primary open angle glaucoma. Arch Ophthalmol 1992;110:1769–1778. Copyright 1992, American Medical Association.) B. Trabecular meshwork, primary open-angle glaucoma. Trabeculectomy specimen from patient with primary open-angle glaucoma shows decreased cellularity of trabecular endothelium and fusion of beams in inner meshwork. These changes may be artifactitious. (PAS ×250)
CLASSIFICATION OF THE GLAUCOMAS
The glaucomas are classified into developmental, primary or idiopathic, and secondary types. Primary and secondary glaucomas are subclassified into open-angle and closed-angle variants depending on whether the angle is open or closed. Angle-closure glaucoma is marked by the apposition or adherence of the peripheral iris to the trabecular meshwork (Fig. 8-6). Developmental glaucomas present in infancy or childhood and may be inherited or are associated with other ocular anomalies or systemic disorders.
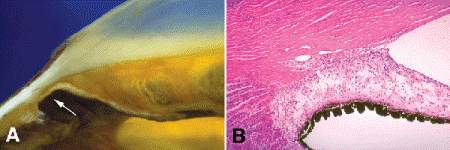
Fig. 8-6. A. Peripheral anterior synechia. Arrow points to area where peripheral iris adheres to trabecular meshwork and posterior cornea. The anterior iridic surface is flattened by a subtle neovascular membrane. B. Peripheral anterior synechia. The peripheral iris adheres to the inner surface of the trabecular meshwork, blocking the outflow of aqueous humor. A neovascular membrane flattens the anterior iridic surface. (H&E ×25)
Developmental Glaucoma
Developmental glaucomas are caused by developmental abnormalities or dysembryogenesis of the aqueous outflow pathways. Primary congenital glaucoma is a bilateral disorder that is often inherited as an autosomal recessive trait. Autosomal recessively inherited congenital glaucoma is caused by mutations in the cytochrome P4501B1 gene (CYP1B1) on chromosome 2 (2p22-p21). Forty percent of cases are present at birth and eighty-six percent become evident during the first year of life. Affected infants often have light sensitivity (photophobia), blepharospasm, and tearing and may be misdiagnosed as having nasolacrimal duct obstruction. Ocular enlargement (buphthalmos or “ox eye”) is the clinical hallmark of congenital glaucoma. Elevation of intraocular pressure only causes ocular enlargement during childhood when the sclera is relatively thin and elastic. Corneal enlargement and ectasia of limbal tissues is especially striking (Fig. 8-7A). As the cornea stretches, Descemet membrane may rupture spontaneously, causing corneal edema and opacification. Old healed ruptures in Descemet membrane in patients with congenital glaucoma are called Haab striae (Fig. 8-7). Haab striae usually are oriented horizontally or concentric to the limbus in the peripheral cornea. This distinguishes them from traumatic ruptures caused by obstetrical forceps, which usually are oriented obliquely.
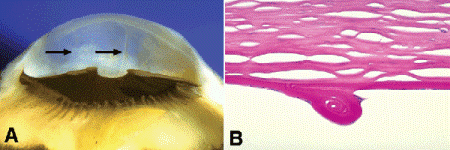
Fig. 8-7. Congenital glaucoma. A. Ridges on back of cornea denoted by arrows are healed ruptures in Descemet membrane (Haab striae). The cornea is large, the anterior chamber is deep, and the limbal tissues are somewhat ectatic. Depigmentation of ciliary body was caused by a prior cyclodestructive procedure. B. Haab stria, congenital glaucoma. A thickened ridge of hypertrophic coiled Descemet membrane has formed at the site of a rupture caused by corneal enlargement. Intrinsically elastic, Descemet membrane often coils up when lacerated or ruptured. (PAS ×100)
Hypothetical mechanisms involved in the pathogenesis of congenital glaucoma include an imperforate mesodermal sheet covering the trabecular meshwork called Barkan membrane, congenital absence of Schlemm canal, and persistence of a fetal angle configuration. Histopathologically, the fetal angle is characterized by anterior insertion of the iris root and ciliary processes, the presence of mesenchymal tissue in the angle, and continuity of ciliary muscle fibers with trabecular beams. The anterior chamber usually is quite deep in eyes with congenital glaucoma. The angle is open gonioscopically, and there is a high insertion of the iris root.
Developmental glaucoma occasionally occurs in association with other ocular abnormalities or congenital syndromes including aniridia, the Axenfeld-Rieger syndrome, and Peters anomaly. Glaucoma also complicates von Recklinghausen neurofibromatosis (NF I) and Sturge-Weber syndrome, particularly if the upper eyelid is involved by the hamartomatous process: a plexiform or diffuse neurofibroma in NF-1 or a nevus flammeus in Sturge-Weber syndrome (Fig. 2-8). Hamartomatous infiltration of the angle may produce a distinctive gonioscopic appearance in neurofibromatosis. The angle is blanketed by a uniform layer of tan tissue which obscures normal trabecular landmarks. Glaucoma and cataract occur concurrently in Lowe syndrome. Other syndromes that may have congenital glaucoma include Gregg congenital rubella syndrome, Stickler syndrome, Hallermann-Streiff syndrome, Hurler syndrome, Turner syndrome, and trisomies 21 and 13.
Primary Open-Angle Glaucoma
Primary or idiopathic open-angle glaucoma (POAG) is the most common type of glaucoma and affects an estimated 1% to 3% of the population. POAG is an insidious disease that causes asymptomatic painless visual loss. By definition, the angle is open on gonioscopic examination. POAG usually is a bilateral disease, and affected patients frequently have a positive family history. The genetics of POAG are complex; the disorder has been linked to 14 genes, most notably the myocilin (MYOC) gene on chromosome 1. Mutations in myocilin are found in 3% to 5% of patients with adult-onset POAG.
The cause of aqueous outflow obstruction in POAG remains uncertain, but the area of obstruction may be located in the JXT in the deepest part of the trabecular meshwork bordering Schlemm canal. Several pathogenetic theories involve obstruction of the meshwork or JXT by glycosaminoglycans or other abnormal extracellular matrix material. Loss of trabecular endothelial cells could lead to fusion of trabecular beams and decreased porosity of the meshwork (Fig. 8-5B). Electron microscopy has shown that the density of trabecular endothelial cells is decreased in patients with POAG. Trabecular endothelial cell death in patients with mutations in the MYOC gene may be related to the intracellular accumulation of abnormal myocilin. Another study showed that the area of the trabecular culs-de-sac, which provide a major proportion of normal outflow resistance, is markedly reduced in POAG (Fig 8-5A). Other hypothetical pathogenetic mechanisms include abnormalities in the formation of giant vacuoles in the endothelial lining of Schlemm canal, or age-related sclerosis in the scleral spur that impedes posterior uveoscleral outflow.
Primary Closed-Angle Glaucoma
Primary closed-angle glaucoma (acute angle-closure glaucoma or acute congestive glaucoma) is caused by functional apposition or blockage of the trabecular meshwork by the peripheral iris. The resultant acute rise in intraocular pressure produces major symptoms including severe ocular pain, headache, and gastrointestinal symptoms (nausea and vomiting) caused by a vagal oculogastric reflex. The involved eye is injected and classically has a fixed, dilated pupil during an acute attack of closed-angle glaucoma. The vision usually is diminished by corneal epithelial edema evident clinically as “bedewing,” or possibly by posterior segment ischemia. Primary closed-angle glaucoma usually is unilateral and classically occurs in hyperopic (“far-sighted”) patients whose small eyes have shallow, crowded anterior chambers. Primary closed-angle glaucoma is extremely rare in myopes (near-sighted individuals) and younger patients less than age 40. Progressive growth of the lens or development of an intumescent cataract can precipitate an acute attack of closed-angle glaucoma in elderly patients (phacomorphic glaucoma). Acute angle-closure glaucoma is more prevalent in certain racial groups (e.g., Asians and Inuits) and often occurs in nanophthalmic eyes that are markedly hyperopic and prone to develop exudative ciliochoroidal detachment. Most patients with angle closure have an asymptomatic course and do not suffer acute attacks. Quigley has hypothesized that disturbed physiological mechanisms contribute to angle closure and angle-closure glaucoma in many cases. Such factors include diminished loss of iris volume during pupillary dilation and expansion of choroidal volume.
Functional pupillary block is involved in the pathogenesis of primary closed-angle glaucoma. When the pupil is mid-dilated, the iris pigment epithelium near the pupil is pressed firmly against the anterior surface of the lens, impeding the flow of aqueous humor into the anterior chamber. The pupillary block is functional because actual adhesions between the iris and lens called posterior synechiae have not formed. Continual production of aqueous humor behind the iris produces a pressure gradient that bows the peripheral part of the iris forward, obstructing the trabecular meshwork. If this functional papillary block is not relieved expeditiously, permanent adhesions between peripheral iris and trabecular meshwork called peripheral anterior synechiae eventually develop. Acute closed-angle glaucoma is cured by making a full-thickness hole in the iris. This equalizes the pressure in the anterior and posterior chambers and allows the iris to fall back into its normal position. The iridotomy usually is performed with a surgical laser.
High levels of intraocular pressure can cause permanent damage to anterior segment structures during an attack of acute closed-angle glaucoma. Ischemic in nature, these changes persist as stigmata of a prior “acute attack” and include permanent dilation and unreactivity of the pupil caused by necrosis of the sphincter muscle, patchy atrophy of the iris stroma, and small grayish anterior subcapsular lens opacities called glaukomflecken. Glaukomflecken probably represent focal areas of lens epithelial necrosis and cortical degeneration.
Secondary Closed-Angle Glaucoma
Secondary glaucomas are caused by concurrent ocular or systemic disease. Both closed-angle and open-angle varieties of secondary glaucoma occur. Many blind glaucomatous eyes examined in the ophthalmic pathology laboratory have secondary closed-angle glaucoma. Secondary closed-angle glaucoma is characterized by the formation of permanent adhesions between iris and trabecular meshwork called peripheral anterior synechiae (Figs. 8-6–8-9). There are many causes of secondary closed-angle glaucoma. Permanent peripheral anterior synechiae can form in untreated primary closed-angle glaucoma and often develop in the late stages of retinopathy of prematurity or persistent hyperplastic primary vitreous (PHPV). Posterior segment tumors usually cause secondary closed-angle glaucoma by stimulating iris neovascularization or by a pupillary block mechanism.

Fig. 8-8. Iris bombé. Pupil is secluded by 360-degree posterior synechiae. Peripheral iris is bowed anteriorly forming broad secondary peripheral anterior synechiae.
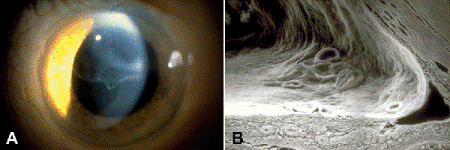
Fig. 8-9. Epithelial downgrowth. A. Slit lamp discloses sheet of corneal epithelium on posterior corneal surface superiorly. B. SEM shows sheet of surface epithelium introduced by trauma lining the posterior cornea, trabecular meshwork, and anterior surface of the iris. The epithelial membrane flattens the anterior surface of the iris. (SEM ×80)
The role of functional pupillary block in primary closed-angle glaucoma has been discussed earlier. Pupillary block also is important in several types of secondary closed-angle glaucoma. Inflammatory adhesions between the pupillary part of the iris and the anterior lens capsule called posterior synechiae readily form in the sticky, fibrin-rich milieu of iritis or iridocyclitis. The entire circumference of the pupil may become firmly bound to the lens (seclusio pupillae), totally blocking the flow of aqueous humor into the anterior chamber. The elevated pressure in the posterior chamber bows the peripheral part of the iris forward (iris bombé) and blocks the trabecular meshwork secondarily (Fig. 8-8). Cycloplegic medications such as atropine or scopolamine relieve the pain of pupillary and ciliary spasm in uveitis and help to prevent posterior synechiae by dilating the pupil. The lens or vitreous can also block the pupil. Anterior displacement of the microspherophakic lens in Weill-Marchesani Syndrome readily occludes the pupil, causing pupillary block glaucoma (Fig. 7-15B). Pupillary block glaucoma also occurs in patients with traumatic or heritable lens dislocation. Prophylactic peripheral iridectomies were performed routinely during intracapsular cataract surgery to prevent postoperative blockage of the pupil by the anterior face of the vitreous. Pupillary block caused by anterior movement of the lens-iris diaphragm is a common cause of secondary closed-angle glaucoma in eyes that have large posterior segment tumors or extensive bullous retinal detachments.
Several clinically important types of secondary closed-angle glaucoma are caused by the proliferation of cells on anterior chamber structures. These secondary proliferative glaucomas include epithelial downgrowth or ingrowth caused by proliferation of ocular surface epithelium after surgical or nonsurgical trauma, the iridocorneal endothelial (ICE) syndrome caused by proliferation of abnormal corneal endothelial cells, and neovascular glaucoma (NVG) caused by iris neovascularization. Epithelial downgrowth is discussed in Chapter 3 (Figs. 4-15A and 8-9).
Neovascular Glaucoma
Many blind painful eyes accessioned by ophthalmic pathology laboratories have NVG (Figs. 8-10–8-12). Peripheral anterior synechia formation in NVG is caused by the proliferation of fibrovascular tissue on the anterior surface of the iris and angle. Angiogenic factors such as vascular endothelial growth factor (VEGF) produced by ischemic parts of the retina or intraocular tumor cells stimulate the iris neovascularization. VEGF levels in the aqueous humor are significantly increased in NVG. Clinically, conditions most commonly associated with NVG include retinal vein and artery occlusions, proliferative diabetic retinopathy, and intraocular tumors, particularly retinoblastoma.
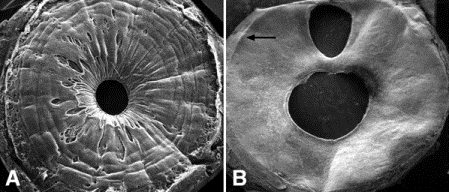
Fig. 8-10. Neovascular glaucoma. A. Scanning electron microscopy of normal iris shows collarette, crypts, and contraction furrows. B. Neovascular glaucoma. Neovascular membrane flattens and effaces normal architecture of anterior iridic surface. A peripheral iridectomy is present. Peripheral ridge (arrow) marks site of ruptured anterior synechia. (A. SEM ×10, B. SEM ×10)
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


