2 Genetics of Hearing Loss Hearing loss is an extremely common sensory deficit that affects communication in nearly 70 million people worldwide. In the United States, one in 1000 newborn infants has severe to profound hearing loss, and one to two in 1000 are born with a lesser but clinically significant bilateral or unilateral hearing loss.1 The widespread establishment of newborn audiologic screening programs now allows the early recognition of hearing loss. Appropriate therapeutic and educational intervention in these children allows them to achieve their full developmental, language, and academic potential.2,3 More than 100 genes have been identified that cause hearing loss, and it seems likely that nearly 1% of all the genes in the genome, or about 300 genes, could ultimately be identified as causes of hearing loss. This chapter highlights some of the significant advances in this rapidly evolving field as they relate to the provision of optimal care for individuals with hearing loss, and discusses future directions for the diagnosis and management of hearing loss. Hearing loss is an etiologically heterogeneous trait with many recognized environmental and genetic causes. Environmental causes still common in Western countries include congenital cytomegalovirus (CMV) infection, prematurity, and ototoxic drug use. Certain environmental causes such as congenital rubella and bacterial meningitis are less common in the United States, but continue to be a leading cause of hearing loss in developing nations. Genetic factors account for at least half of all cases with profound hearing loss.4 In nearly 30% of the genetic cases one can identify a recognizable pattern of clinical features involving secondary organ systems. More than 300 of these syndromic forms of deafness have been described to date.5 In the remaining 70%, hearing loss is the sole manifestation and often may not even be associated with a history of deafness in the family. This is commonly the case with autosomal recessive transmission, which accounts for 80% of all genetic cases; another 15% is transmitted as an autosomal dominant trait; and 1 to 2% each are associated with X-linked and mitochondrial transmission. The frequency of mitochondrial hearing loss exhibits considerable variation among populations. Less is known about the etiology of mild to moderate as well as unilateral hearing loss. Advances in strategies for mapping and ultimately cloning genes for deafness, along with the completion of the Human Genome Project, the establishment of a human fetal cochlear complementary DNA (cDNA) library, and the exploitation of murine models for hearing loss, have contributed to the accelerated discovery of a growing number of genes for deafness.6,7 To date, 87 loci for nonsyndromic deafness have been mapped, and 42 genes have been identified of which 21 are for recessive, 20 for dominant, and one for X-linked hearing loss.8 Mutations involving two mitochondrial genes, the 12SrRNA and tRNA ser (UCN), result in nonsyndromic hearing loss (NSHL). The nomenclature used to denote the dominant, recessive, and X-linked loci is DFNA, DFNB, and DFN, respectively. For example, DFNB1 indicates the first recessive locus for NSHL mapped by Guilford et al9 in 1994, with GJB2 (Gap Junction beta-2) and GJB6 (Gap Junction beta-6) being two genes identified in this region on the long arm of chromosome 13. The identification of genes involved in hearing loss is gradually unraveling their function, their interactions with one another, and their role in the complex process of sound perception.10,11 Several fascinating observations have emerged from these studies, such as the occurrence of digenic hearing loss,12 the presence of modifier genes affecting the deafness phenotype,13 the involvement of the same gene in both dominant and recessive NSHL (at least seven genes), and mutational heterogeneity in a single gene that can lead either to syndromic or nonsyndromic hearing loss.14 Nearly 400 syndromes have been described in which the presence of hearing loss and specific clinical findings permits the diagnosis of a recognizable pattern or a syndromic form of hearing loss. The clinical finding of hearing loss is quite variable in severity and in the age of onset in these entities, and occasionally may not be a constant feature. Although the vast majority of these syndromes are relatively rare, a few are more common or should be considered in all individuals with hearing loss because of their clinical implications. Table 2–1 lists the clinical features of several important forms of syndromic deafness whose recognition can have great clinical relevance. More comprehensive reviews and information for these conditions can be obtained on several Internet sites and reviews in the literature.5,7,8,15,16 In the large proportion of genetic hearing loss (70–80%) that is nonsyndromic, deafness is the only identifiable clinical finding. Although a family history of hearing loss in a sibling or other family member (multiplex cases) can help determine the genetic mode of inheritance, recent studies indicate that in nearly 15 to 20% of patients with hearing loss, there may be no history of deafness in any relatives. These simplex cases are often erroneously referred to as “sporadic” or nongenetic cases that are therefore associated with a negligible recurrence risk. In fact, simplex cases include “chance-isolated genetic cases” typically associated with a 25% recurrence risk in addition to true sporadic cases caused by environmental factors that are associated with a very low recurrence risk. It is typically difficult to distinguish the different genetic forms of deafness based solely on the audiologic characteristics, but when there are multiple affected family members, the age of onset, progression, and audiologic findings are often remarkably similar. In many cases, a single large family has been instrumental in the identification of a novel deafness locus, and in some cases these original families have remained unique examples of the mutation in question. In view of the large number of loci already identified, it was an astonishing surprise to find one particular locus, DFNB1, involving mutations in two connexin genes mapped to this region, to account for nearly 50% of all recessive deafness.17 An attempt has also been made to establish genotype and phenotype correlations between the type of hearing loss and a genetic locus; for example, mutations in the Wolframin gene (WFS1) on chromosome 4 are associated with dominantly inherited low-frequency NSHL,18 whereas Meniere-like symptoms can be seen with dominant NSHL with mutations in the COCH gene.19 As new genes for deafness are mapped and cloned, their characterization has allowed some inference about their expression in the ear, its function, and an understanding of the pathogenesis of hearing loss when these units are altered. Table 2–2 summarizes and presents a succinct view of some of these genes and the molecular basis of hearing with a brief clinical overview as we know it today. A few forms of genetic deafness with the greatest clinical relevance due to their frequent occurrence, availability of clinical molecular testing, potential for prevention, or a characteristic phenotype are discussed in greater detail below. The connexins are a family of genes that code for subunits of gap junction proteins. Gap junctions are hexameric structures formed by the docking of six connexon units on adjacent cell surfaces. These gap junction channels allow the free flow of ions and small molecules across membranes between two adjacent cells. Both α and β connexins have been described, which are designated by numbers that refer to the molecular weight of the protein.20 In 1997 Kelsell et al21 identified connexin 26 to be the gene responsible for hearing loss in families with DFNB1, mapped to 13q12. Connexin 26 gene encodes the Gap Junction β2 protein (GJB2) and is expressed in the inner ear in the fibrocytes and supporting cells as well as in the stria vascularis. These gap junctions are instrumental in recycling and maintaining the high potassium content of the endolymph, which is critical for generating the appropriate electrical sound potentials.10 As noted previously, quite contrary to the belief that hearing loss would be determined by rare genes at a large number of loci, mutations in the GJB2 gene are responsible for 50% of recessive deafness and 20% of all profound deafness in childhood.22–24 A one base pair (bp) deletion of the guanine nucleotide in a string of six G’s between bp 30 and 35 in the GJB2 gene is the most common pathologic change and accounts for 70% of all mutations.25 More than 100 different mutations spanning the entire protein have been reported to be associated with hearing loss.26 Some of these mutations are specific to particular ethnic groups; for example, 35delG in Caucasians from Western Europe, 167delT in Ashkenazi Jews, 235delC in East Asians, and the R143W variant in Ghana.27 The majority of individuals with two pathogenic mutations in GJB2 have severe to profound prelingual hearing loss, which is typically present at birth.28,29 However, no systematic studies have been done to date to assess this, and anecdotal reports of infants with GJB2 mutations who passed their newborn audiologic screen but later developed hearing loss may suggest that the phenotype may not always be fully penetrant at birth. Certain sequence variants such as V37I and L90P are associated with a milder hearing loss. The pathogenic role of other changes, such as the M34T mutation, remains controversial.30,31GJB2 deafness is not associated with inner ear malformations or vestibular findings. However, a few GJB2 mutations also cause dermatologic abnormalities such as palmoplantar hyperkeratosis, which is seen with the dominant G59A allele,32 mutilating keratoderma (Vohwinkle syndrome) with the D66H allele,33 and the keratoderma-ichthyosis-deafness (KID) syndrome with three other dominant alleles.34 The frequency of heterozygote carriers of the GJB2 mutation in the midwestern U.S. population is reported to be 3.5%.35 Recently, del Castillo et al36 reported the presence of a 342-kilobase (kb) deletion spanning the GJB6 (Cx30) gene in-trans with a 35delG mutation in the GJB2 gene in 66% of deaf GJB2 heterozygotes in Spain. Interestingly, GJB6 lies 35 kb upstream of the 5′ end of GJB2 on 13q11.2 within the DFNB1 locus. In a large repository of 737 deaf probands from the U.S., 2.5% had the 342-kb deletion, and it was found in 15.9% of deaf GJB2 heterozygotes.24 Interestingly, the hearing loss in deaf probands with the 342-kb deletion and a mutation in the GJB2 gene was more severe than in deaf individuals with two GJB2 mutations. This observation, although based on a relatively small sample size, supports the idea that digenic inheritance involving defects in both connexin proteins causes the hearing loss. Several other β connexins such as connexin 30 and 31 have been reported to result in hearing loss in single families.37,38 Connexin 32 results in an X-linked form of Charcot-Marie-Tooth neuropathy with hearing loss.39 This particular form of deafness leads to a distinctive audiologic profile of low-frequency hearing loss (LFHL). The hearing loss begins early in life and shows little or no progression, and was first described in an American family in 1968,40 mapped to 4p16.3 in 1995 by Lesperance et al,41 and ascribed to mutations in the Wolfram syndrome I gene in 2001.18 Two other families, one Dutch and another French-Canadian, with a similar phenotype except for mild progression of hearing loss, were mapped to the same region and designated DFNA14 and DFNA38, respectively.8 Both these families were subsequently shown to have allelic mutations at the same WFS1 locus,42 and mutations at this gene are now thought to be the commonest cause of LFHL. Interestingly, Wolfram syndrome is a recessive disorder characterized by both diabetes insipidus and mellitus, optic atrophy, progressive high-frequency hearing loss, and a variety of neuropsychiatric symptoms such as depression, violent behavior, suicidal tendencies, seizures, ataxia, and retardation.43 The disorder is referred to by the acronym DIDMOAD, which stands for diabetes insipidus, diabetes mellitus, optic atrophy, and deafness (Online Mendelian Inheritance in Man No. 222300).16 More than 60% of individuals with DIDMOAD carry inactivating mutations in the WFS1 gene, affecting the transmembrane domains of the encoded protein, resulting in a loss of function. In contrast, all of the mutations reported in families with dominant LFHL, with the exception of one mutation in DFNA6/14, have missense substitutions in the fifth intracellular domain of the WS1 gene.44 Although the majority of dominant LFHL results from mutations in the WS1 gene, a second locus, DFNA1, is also associated with LFHL, but the phenotype exhibits rapid progression to involve all frequencies in later life. To date, this form of deafness has been identified in only a single large Costa Rican family and was shown to result from a mutation in the human homologue of the diaphanous gene in Drosophila.45
Epidemiology of Hearing Loss
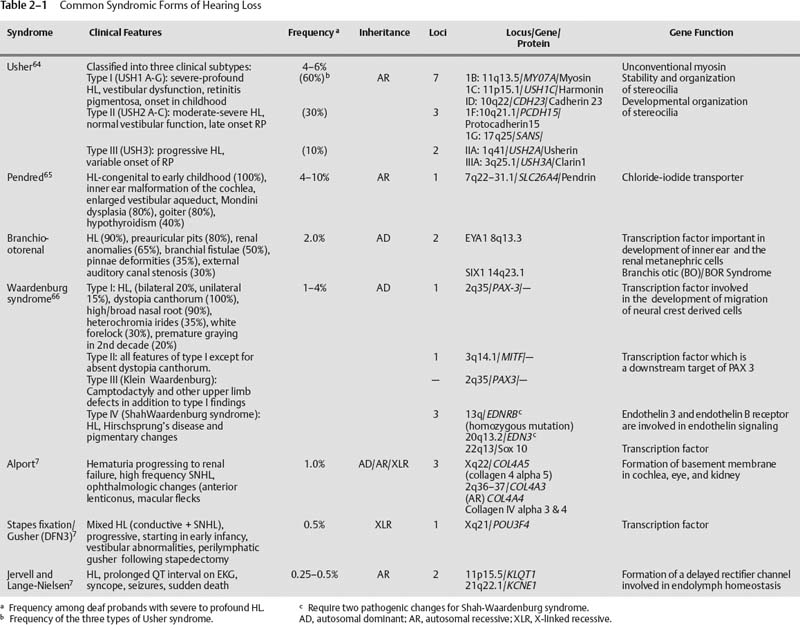
Common Syndromes Associated with Hearing Loss
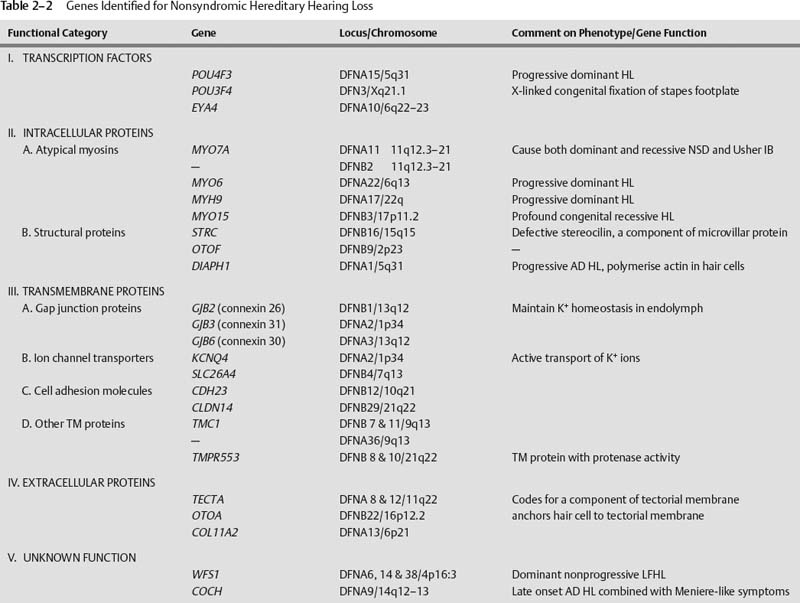
Nonsyndromic Hearing Loss
Connexin Deafness
Dominantly Inherited Low-Frequency Hearing Loss (DFNA6, 14, and 38)
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


