Chapter 10 Genetics and pediatric ophthalmology
Background
In developed countries, half of the conditions causing childhood blind and partially sighted registration are genetic,1–3 a figure that is likely to be underestimated. In many developing countries where childhood visual disability is significantly commoner, genetic conditions also represent an important group contributing to childhood blindness.1,4–6 “Genetic” conditions referred to in this context are monogenic, (Mendelian) conditions. Since many issues regarding diagnosis and counseling apply to the group as a whole, this allows a common approach to clinical management. However, the substantial genetic contribution to common diseases, i.e. the delineation of genetic variants in the complement pathway as contributors to AMD, and normal quantitative traits (corneal thickness, optic nerve size) underlines the observation that molecular genetic discoveries are not limited to Mendelian disease.
The study of inherited ocular disease represents one of the successes of modern molecular genetics, from the description of linkage of xlRP7 to the identification of the first adRP gene encoding rhodopsin.8 The Human Genome Project has accelerated the understanding of the molecular basis of human genetic disease. Now, over 200 gene loci and 150 genes have been described underlying human monogenic retinal disorders, implying a level of complexity unsuspected 20 years ago (http://www.sph.uth.tmc.edu/retnet/).
Mendelian inheritance
Autosomal dominant inheritance (Fig. 10.1)
New mutations
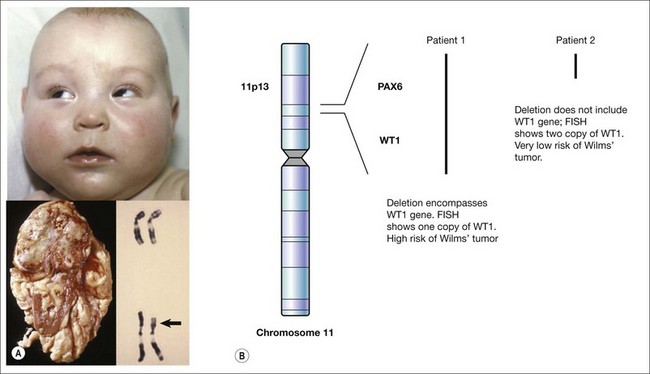
The exact nature of a de novo mutation is difficult to predict – for cases of sporadic aniridia, a deletion can remove other neighboring genes. This is seen in WAGR syndrome where a deletion causes Wilms’ tumor, aniridia, genitourinary abnormalities, and intellectual retardation.9–11 This is termed a contiguous gene syndrome. It is for this reason that patients with sporadic aniridia require either renal ultrasound screening or molecular evidence that the Wilms’ tumor gene, WT1, is unaffected by the new mutation (Fig. 10.2).
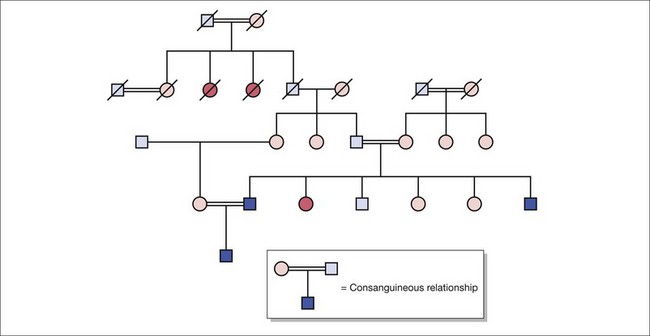
Fig. 10.2 Pedigree construction illustrating autosomal recessive inheritance in the presence of consanguinity.
Autosomal recessive inheritance (Fig. 10.3)
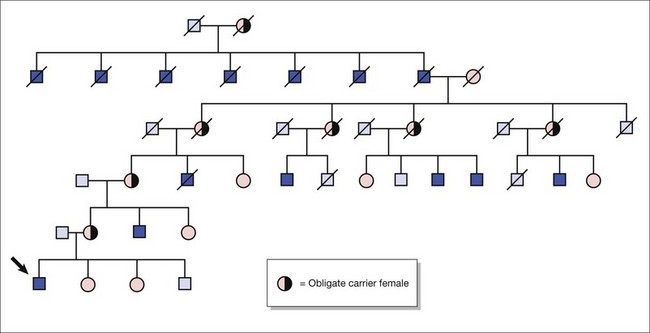
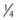 . Unaffected children have a
. Unaffected children have a 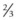 risk of being carriers.
risk of being carriers.Calculating carrier frequencies in the general population is complex. For inherited eye conditions, where one condition may be caused by many different genes (e.g. retinal dystrophy), accurately predicting the frequency of any one of those genes in a given population is often not possible. For Stargardt’s disease with an estimated disease frequency of 1 in 10 00012 and a carrier frequency of 1 in 50, the risk to the offspring of an affected individual and their children is low (~1% and 0.65%, respectively).