Most genetic hearing disorders are transmitted by an autosomal recessive mode of inheritance, and of the hearing disorders presenting in childhood, approximately 80% are inherited recessively. In most patients, hearing loss is nonsyndromic, but 10% to 20% of patients display syndromic disorders. The identification of recessive syndromes involving hearing loss, thus, necessitates a diligent search for other syndromic components. Because of the asymptomatic status of heterozygous carriers of a recessive gene and the 25% inheritance risk, it is often difficult to distinguish between nongenetic disorders and those that are recessively inherited. In both clinical situations, a single affected child generally presents in a family with no known history of hearing loss.
Nonsyndromic Recessive Disorders
To date, linkage studies have identified at least 85 loci for autosomal recessive nonsyndromic hearing loss (ARNSHL) (
2). These loci are given the prefix DFNB; DFN signifies deafness, whereas B signifies a recessive mode of inheritance. Thirty seven causative ARNSHL genes have been identified (
Table 102.1). DFNB1 accounts for about one half of all ARNSHL and, as such, is among the most active areas of clinical research. As implied by its designation,
DFNB1 was the earliest autosomal recessive gene locus to be successfully mapped and characterized. The gene at the DFNB1 locus is gap junction beta 2 (
GJB2), which produces a protein called connexin 26 (
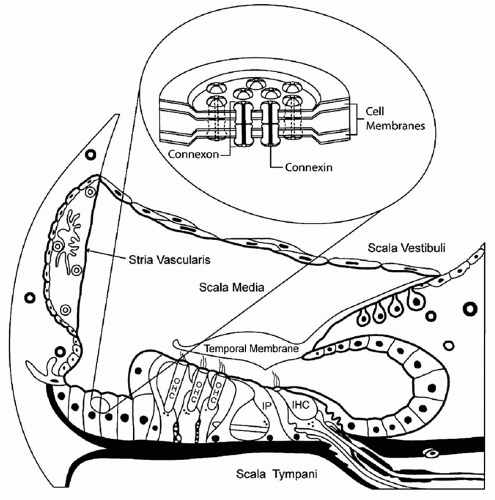
1). Connexins comprise a family of gap junction proteins that function as intercellular channels. These channels allow ions and low molecular weight molecules to travel from cell to cell (
Fig. 102.2). The supporting cells of the organ of Corti express several gap junction proteins, including connexin 26. Although the function of connexin 26 in the inner ear has not been conclusively determined, it is thought to be involved in potassium recycling. In the normal ear, a sound stimulus causes depolarization of hair cells, which is mediated by the influx of potassium ions. These ions are thought to transverse through connexin molecules back to the stria vascularis. From the stria vascularis, ions are actively pumped back into the endolymph to reset its electrical gradient (
Fig. 102.2).
DFNB1 is estimated to cause up to 50% of all ARNSHL in many populations. Of patients presenting with hearing impairment of 70 dB or greater, nearly 40% have DFNB1; of those with a mild to moderate hearing loss, only 10% to 15% have this gene (
3,
4).
GJB2 mutations have been found in patients from many different ethnic backgrounds, and specific mutations vary with patient ethnicity. The deletion of a guanine at nucleotide position 35 of the gene (35delG) is one of the most common mutations that results in autosomal recessive hearing impairment, particularly in patients of European descent. The carrier frequency for the 35delG deletion in Europe is 1 in 50. A deletion of a thymine at position 167 (167delT) is often present in the Ashkenazi Jewish population, with a carrier frequency of 1 in 25. A 235delC mutation occurs more frequently in Asians. Among the white population in the United States, the frequency of heterozygous carriers of mutations in
GJB2 is approximately 1 in 40. This carrier frequency is almost as common as that in cystic fibrosis, which has a carrier frequency of approximately 1 in 24. The high frequency of mutations and the small size of this gene make screening and molecular genetic diagnosis feasible.
Additionally, a unique genetic mutation has been found in the
GJB6 gene. This mutation appears to present in carriers of
GJB2 mutations (double heterozygote state). A large deletion (170 to 340 kilobases [kb]) upstream and including the 5′ portion of the
GJB6 gene has been found to cause severe hearing loss (
5). Interestingly,
GJB2 and
GJB6 are located in close proximity on chromosome 13, and this deletion may interfere with a common promoter element for the two genes (locus control region). Approximately 10% of patients with hearing loss who have one abnormal
GJB2 allele also have the
GJB6 deletion (
6).
GJB2 mutations can create altered protein expression, subcellular localization, or abnormal functionality of the connexin complex.
GJB2 mutations cause a wide range of
hearing loss phenotypes, ranging from mild to profound hearing impairment. These mutations can be nontruncating or truncating. The type of mutation can produce varying levels of hearing loss. For example, a biallelic truncating mutation produces a severe to profound hearing loss in nearly all patients (e.g., 35delG/35delG). In contrast, the presence of one nontruncating mutation (e.g., V37I) commonly causes a milder form of hearing impairment (
2).
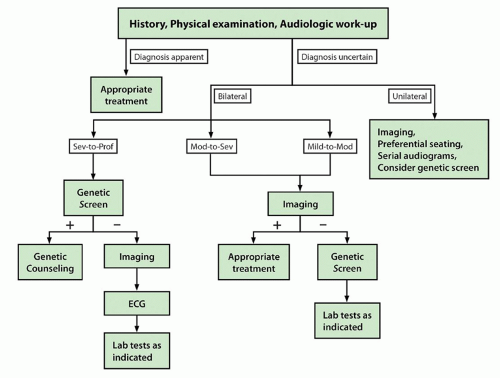
The clinical significance of DFNB1 mutations lies in diagnostic evaluation and case management. Genetic testing is now routinely part of the algorithm in the evaluation of hearing loss (
7). Genetic testing should be the first line of evaluation of children with bilateral ARNSHL of 60 dB or greater (
Fig. 102.3). For those with unilateral or mild bilateral hearing impairment, genetic testing should be considered after temporal bone imaging is performed. If the imaging study is normal, genetic testing is then appropriate. If the imaging study demonstrates enlarged vestibular aqueduct or Mondini deformity,
SLC26A4 (Pendred) testing should be performed. New genetic platforms will allow rapid screening for multiple genes at one time (e.g., “Ear-gene,” Cincinnati Children’s Hospital Medical Center, Cincinnati, OH). Recent data have shown that gene chip platforms are highly accurate and will likely become instrumental in diagnostic evaluation of sensorineural hearing loss (SNHL) (
8).
The prognosis for children with hearing loss related to
GJB2 is variable and has been shown to correlate with the genotype. This is particularly important in light of new hearing screening programs that are increasingly detecting hearing loss in very young children. Genetic information can assist in predicting the actual hearing phenotype, which is valuable considering the emphasis on early (age 12 months) cochlear implantation. For example, if a nontruncating mutation is present, a child would be more likely to have mild hearing loss. Conversely, the presence of biallelic truncating mutations in a child in whom an auditory brainstem response (ABR) test at 1 month of age showed severe hearing loss would lead the practitioners to recommend early aggressive treatment. A high likelihood would exist of recommending cochlear implantation to ensure the child’s ability to communicate verbally. Interestingly, research indicates that patients with DFNB1-related hearing loss who meet criteria for cochlear implantation have better outcomes than those with congenital hearing loss from other causes (
9). Such findings emphasize that genotype can have clinical significance, and further study aimed at elucidating this particular association is ongoing.
Most other ARNSHL genes have been identified from isolated consanguineous families, and the prevalence of mutations in these genes in the general population has yet to be determined. A technology now being developed (resequencing microarray), however, will enable the rapid reading of a large amount of DNA.
Another ARNSHL gene worthy of note is otoferlin (
OTOF). This gene was originally found to be the cause of hearing loss in several isolated families (DFNB9) (
Table 102.1). Although the function of
OTOF is not certain, it is hypothesized that this gene is a calcium sensor involved in hair cell exocytosis (
10,
11). Research has shown that in some patients,
OTOF is also responsible for auditory neuropathy (AN) (
12). AN is a unique form
of SNHL characterized by absent ABR waveforms, present otoacoustic emissions, and variable behavioral thresholds. The prevalence of AN in patients with SNHL ranges from 0.5% to 15%. The etiology of AN is quite heterogenous with approximately 40% of cases having a genetic basis. It can be autosomal recessive, as in
OTOF; autosomal dominant; X-linked; or syndromic. Some patients with AN also have other peripheral neuropathies or central nervous system disease. The treatment of AN can be challenging, involving the judicious use of FM devices and hearing aids as well as cochlear implantation. A better understanding of the molecular basis of this disease will allow for more accurate diagnosis and a more individualized treatment approach.