Chapter 29 Familial Nonmedullary Thyroid Cancer
Introduction
In 1955, Robinson and Orr published the first report of isolated familial papillary thyroid cancer affecting 24-year-old identical twins.1 Twenty years later, Němec and colleagues described differentiated thyroid cancer without environmental exposures in a mother and son.2 Since that time, numerous reports have been published describing families with thyroid cancer of follicular cell origin without other familial syndromes. Population-based studies have established that individuals with a close relative with thyroid cancer have a five- to ninefold increased risk of developing thyroid cancer themselves. The familial clustering of thyroid cancers of follicular cell origin is now recognized as a discrete entity called familial nonmedullary thyroid cancer (FNMTC), which is characterized by distinct clinicopathologic characteristics. This chapter reviews the epidemiology, classification, clinical features, genetics, treatment, prognosis, and outcomes of FNMTC.
Epidemiology
The incidence of thyroid cancer has doubled since the 1980s, and it is estimated that more than 37,000 new cases will be diagnosed in 2010.3 Approximately 5% of these are medullary cancers that arise from the calcitonin-producing parafollicular C-cells. A familial occurrence and association with multiple endocrine neoplasia (MEN) types 2A and 2B have been well described for medullary thyroid cancer.3 The remaining 95% of thyroid cancers are nonmedullary cancers that arise from thyroid follicular cells with four predominant histologic subtypes: papillary (85%), follicular (11%), Hurthle (3%), and anaplastic (1%).4,5 Although the majority of cases are sporadic, numerous reports have described familial clustering, now known as FNMTC, which accounts for 3.8% to 6.2% of all thyroid cancer cases.6–8
Classification of Familial Disease
Currently, FNMTC is defined by the presence of well-differentiated thyroid cancer of follicular cell origin in two or more first-degree relatives, in the absence of other predisposing hereditary or environmental causes. However, statistical estimates by Charkes suggest that 62% to 69% of cases with only two affected family members may in fact be sporadic. The likelihood of sporadic disease falls to less than 6% in families with three or more affected members.9
FNMTC encompasses a heterogeneous group of nonsporadic diseases and may occur as part of a syndromic complex. FNMTC may occur as a component of a familial syndrome such as familial adenomatous polyposis (FAP), Gardner’s syndrome, Cowden’s disease, Carney’s complex type 1, Werner’s syndrome, and papillary renal neoplasia or may occur in isolation (i.e., without other syndromic association).10–22 McCune-Albright syndrome, Peutz-Jeghers syndrome, and ataxia-telangiectasia (Louis-Bar syndrome) may also be associated with the development of FNMTC, but there are more limited data linking these syndromes with FNMTC.23–25 Known clinical characteristics and genetic causes of some of these syndromes are shown in Table 29-1.
Table 29-1 Syndromes Associated with FNMTC: Clinical Characteristics and Genetic Causes of Syndromes
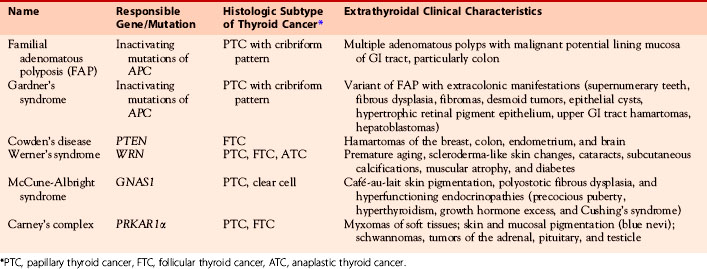
Several epidemiologic studies have documented a five- to tenfold increased risk of thyroid cancer in the first-degree relatives of subjects with nonmedullary thyroid cancer.22,26,27 In a population-based case-control interview study of 159 cases of thyroid cancer with 285 age- and sex-matched controls in Connecticut, the odds ratio of a first-degree relative developing thyroid cancer was 5.2 times that of the general population.26 A Utah population database study demonstrated an increased relative risk (8.6, 95% CI, 4.7 to 13.7) of thyroid cancer in first-degree relatives of thyroid cancer subjects.27 Similarly, a Canadian analysis of 339 patient pedigrees compared to 319 unaffected ethnically matched controls found that 5% of patients with nonmedullary thyroid cancer reported at least one first-degree relative with thyroid cancer.22 A population-based study of the Swedish cancer registry demonstrated that the risk of developing thyroid cancer in a given individual is higher when the affected family member is a sibling and is even higher when the sibling is an affected sister.28 The relative risk of developing papillary thyroid cancer was 3.21 when a parent and 6.24 when a sibling was diagnosed with thyroid cancer. The relative risk was the highest when the two affected siblings were sisters (relative risk 11.9). From an affected mother, the risk was equally high for sons and daughters; however, when either parent was diagnosed with thyroid cancer, the risk for sons was 4.98 (95% CI, 2.13 to 9.86) and for daughters 3.44 (95% CI, 1.96 to 5.60).
A major drawback of population-based studies is that they cannot control for a selection bias or for confounding factors such as environmental exposures (i.e., radiation exposure, iodine deficiency, or excess) that might contribute to a higher risk of thyroid cancer. However, FNMTC has also been reported in large multigenerational kindreds with up to 16 family members affected, where environmental factors were not uniform. These studies of affected familial groups also demonstrate characteristics that appear to be unique as compared to sporadic disease, further supporting the fact the FNMTC is a distinct entity. These include association with second site malignancy, younger age at presentation (30 ± 11), and higher rate of affected males than in sporadic cases. For example, a meta-review of 15 case reports described a median age of 39 years and a male-to-female ratio of 1:2.2.29 FNMTC, unlike sporadic cancers, has been associated with nonthyroid adenocarcinomas. Pal and colleagues found that the incidence of any type of cancer was 38% higher in relatives of patients with thyroid cancer.22 The Utah population database revealed a highly significant association between thyroid tumors with familial clustering and leukemia, breast, prostate, and soft tissue tumors.27 Similarly, other studies have demonstrated an association with breast, kidney, colon, and bladder cancers, and melanoma and lymphoma.30,31 When comparing 47 cases of first- and second-generation of FNMTC patients, Capezzone and colleagues found that offspring show an earlier age at disease onset and have more aggressive disease when compared with their parents, a feature known as genetic anticipation.32 This phenomenon is defined as the occurrence of a genetic disorder at progressively earlier ages and with increased severity in successive generations and is characteristic of inherited neoplasia. These data taken together suggest that FNMTC is a true familial disease rather than chance occurrence of sporadic disease.
Clinical Features
Uchino and colleagues identified 258 patients from 154 families and found that compared to patients with sporadic disease, the FNMTC group had significantly higher rates of coexistent benign thyroid tumors, multicentric tumors, and lymph node metastasis.33 The 30-year rate of recurrence in local lymph nodes was about 40% for FNMTC compared to 20% in the sporadic cases. There was also a higher recurrence rate and worse disease-free survival in this group. There was no difference in age at diagnosis, primary tumor size, rate of extrathyroidal invasion, distant metastasis, or overall survival between the two groups. However, this study diagnosed patients with FNMTC when at least one first-degree relative was affected by thyroid cancer, thereby possibly including cases of sporadic disease as FNMTC.
A retrospective review of 14 patients with FNMTC found a 93% rate of multicentricity and a 43% rate of bilateral disease.30 Patients with FNMTC had higher rates of extrathyroidal invasion in 57% as compared to 5% to 14% in sporadic cases in addition to an increased recurrence rate of 50%.30 Another large cohort study with a multicenter case-matched design found a higher recurrence rate of 44% as compared with 17% in their control group.34 The researchers also found that the median disease-free survival was significantly shorter for patients with FNMTC compared with their matched controls. The number of family members affected with thyroid cancer and distant metastasis was the significant predictor of disease-free survival in familial cases. Perhaps the most compelling study to demonstrate the aggressive nature of FNMTC is the descriptive study by Lupoli and colleagues of 119 patients with papillary thyroid microcarcinoma, of which 7 had FNMTC.35 The rates of tumor multicentricity, bilobar disease, lymph node metastasis, vascular invasion, and recurrence rates were all significantly higher as compared to sporadic cases.
Despite the multiple studies suggesting an aggressive nature of FNMTC, not all investigators have found this to be the case. Ito and associates found no difference in disease-free survival after comparing 273 patients with FNMTC to sporadic cases of papillary thyroid cancer.36 The patients with familial papillary thyroid cancer had a higher rate of multicentricity but no difference in overall prognosis as compared to sporadic cases. These workers also found six families with presumptive familial follicular thyroid cancer, a prevalence of 1.9%.37 Aggressive features such as extrathyroidal invasion, tumor size, and lymph node and distant metastases as well as overall prognosis did not differ from those with sporadic follicular thyroid cancer. A meta-review conducted by Loh also did not demonstrate a more aggressive biology when evaluating rates of multicentricity, local invasion, lymph node and distant metastases, and recurrence rates.29
Tumor Histology
Familial papillary thyroid cancer is the most common type of FNMTC followed by Hurthle cell carcinoma then follicular thyroid cancer. Familial papillary thyroid cancer is characterized by multicentric tumors without any distinguishing pathologic features and multiple adenomatous nodules with or without oxyphilia.38–41
Familial Adenomatous Polyposis (FAP)
Thyroid cancer associated with FAP is usually bilateral and multifocal. Papillary thyroid cancer occurs with a frequency of about 10 times greater than that expected for sporadic cases in patients with FAP.38 The histologic features are different from sporadic tumors with the characteristic cribriform pattern having solid areas and a spindle cell component, most often associated with marked fibrosis. The cribriform-morular variant is a rare subtype of papillary thyroid cancer in this patient population accounting for approximately 0.1% to 0.2% of cases.14
Cowden’s Disease
Multiple adenomatous nodules are a characteristic finding in this syndrome.42 Gross examination reveals firm yellow-tan well-circumscribed nodules diffusely involving the thyroid gland. Microscopically, they are well-circumscribed nonencapsulated solid cellular nodules sharing features similar to follicular adenomas. Some nodules may have a discontinuous rim of fibrous tissue simulating a capsule. Follicular adenomas are common and usually multiple in this syndrome. Follicular carcinomas may also arise from a preexisting follicular adenoma. Papillary thyroid cancer has rarely been associated with this entity.42
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


