Eye Movements
R. John Leigh
Janet C. Rucker
A grasp of the properties of eye movements is indispensable to practicing ophthalmologists. Abnormal eye movements often provide diagnostic clues and sometimes cause visual complaints that the ophthalmologist is well qualified to manage. Thanks to an ongoing basic research effort, it is now possible to define the neural substrate for normal eye movements and to provide credible explanations for most gaze disorders and their visual consequences.1
The overall purpose of eye movements is to guarantee that our view of the world is clear and stable. In order to provide optimal viewing conditions during a variety of visual tasks, several functional classes of eye movements have evolved (Table 1).2 Although the visual system can tolerate some slip of images on the retina,3 if this slip exceeds a threshold that is determined by the spatial frequency of the object under view (approximately 5 degrees per second for higher spatial frequencies),4 then visual acuity declines and an illusion of movement of stationary objects (oscillopsia) may result. Normally this is prevented by gaze-holding eye movements. The first of these is the fixation mechanism, which depends on the visual system’s ability to detect and respond to excessive slip of images on the retina. Visual fixation requires directed visual attention and responds within 100 ms. The second is the vestibulo-ocular reflex (VOR), which depends on motion detectors in the inner ear and generates eye rotations to compensate for head perturbations at short latency (less than 15 ms) so that vision remains clear during locomotion.5,6 Thus, the vestibulo-ocular reflex acts much more promptly than visually mediated fixation. Loss of the vestibulo-ocular reflex caused, for example by aminoglyoside antibiotics, causes oscillopsia during locomotion,7 because the visual system cannot act promptly enough to compensate for the head vibrations that occur with each footstep. During sustained (lower frequency) head movements, however, the visual system can contribute to holding gaze steady by generating optokinetic eye movements. When the eyes are turned out to an eccentric position in the orbits, then the brain must take into account the mechanical pull of the orbital suspensory tissues (such as Tenon’s capsule) and program movements accordingly. This eccentric gaze-holding function, depends on the ability of the brain to generate a tonic contraction of the extraocular muscles; when faulty, the result is gaze-evoked nystagmus, one of the most common abnormalities of eye movements.
TABLE 1. Functional Classes Of Eye Movementsa* | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||
A second group of eye movements is concerned with pointing the fovea at an object of interest. These gaze-shifting eye movements are more recently evolved than the gaze-holding mechanisms, and are absent in animals lacking a well developed fovea, such as the rabbit. Saccades are rapid movements by which we place the image of an object of interest on fovea. They are important for visual searches of our environment. Saccades can be generated voluntarily, or in response to visual stimuli (presented in the periphery of vision), auditory, somatosensory, or vestibular inputs. The last of these are the quick phases of nystagmus, which reset the eye during sustained gaze-holding movements. Smooth pursuit eye movements hold the image of a moving target close to the fovea, and also allow sustained visual fixation of a stationary target while we are in motion. Vergence eye movements turn the eyes in opposite directions so that the images of a single object can be place simultaneously on both foveas.
It should be realized that under natural conditions there is always interaction and cooperation between different classes of eye movements; thus, vergence is usually associated with fast (saccadic) or slow (pursuit) version movements, because natural gaze shifts are seldom strictly confined to changes in depth (requiring pure vergence) or direction (requiring pure version). For didactic purposes, however, it is useful to separate them. Each functional class of eye movements has distinctive properties that suit it to its specific aims and some of these are summarized in Table 1. Knowledge of these properties will guide the clinical examination. In addition, each functional class of eye movements has a distinct neural substrate, which is discussed below. Familiarity with the neural substrate for each functional class of eye movements aids topological diagnosis.
Our approach will be bottom-up: first to review the extraocular muscles and their innervations, and then examine how neural commands for each functional class of eye movements reaches the ocular motoneurons that reside in the third, fourth, and sixth cranial nerve nuclei. For clarity, we first describe the control of horizontal eye movements, which resides in the pons and medulla, and then control of vertical and torsional eye movements, which depends mainly on the midbrain. We then summarize the cerebellar contributions to control of eye movements, and review cerebral influences on gaze. We conclude by presenting an approach for interpreting nystagmus and saccadic intrusions by applying this scheme for gaze control. As we proceed from extraocular muscles to cerebral cortex, we review normal structure and function and, at each stage, describe classic effects of disease; our account of the latter cannot be comprehensive, and the reader is referred to current texts.1,8,9,10
THE OCULAR MOTOR PERIPHERY: THE EXTRAOCULAR MUSCLES AND THEIR INNERVATION
ANATOMY OF THE ORBITAL CONTENTS AND ROTATIONS OF THE EYES
The eyeball is suspended in the orbit by fascia, the main component of which is Tenon’s capsule. Each eye is rotated by six muscles: four rectus muscles and two oblique muscles. The directions of pulling action of the extraocular muscles depend on the starting position of the eye. The primary action of the muscle refers to the axis around which the eye principally rotates when that muscle contracts; the secondary and tertiary actions refer to the axes around which there are lesser rotations. The pulling actions of each muscle are summarized in Table 2. Concepts of the way that the extraocular muscles attach to the eye and move it have undergone a revolutionary change in the past decade, based on magnetic resonance imaging (MRI) in volunteers and dissection of cadaver orbits.11,12 These studies have demonstrated fibroelastic sleeves for the extraocular muscles that act as pulleys and, for example, stop slide-slip of the horizontal rectus muscles during vertical eye movements. It is well known that the extraocular muscles have two layers, but it now appears that the outer orbital layer inserts into the pulley of the muscle while the inner global layer passes through the pulley to insert onto the globe. Different populations of ocular motoneurons may supply the global and orbital layers, and their proprioception may differ.13 At first sight, such an arrangement raises many complications about the control of eye movements, because it suggests that separate populations of ocular motoneurons will be needed to contract the orbital layer to position the pulley (which is the functional origin of the muscle) and contract the global layer to move the eye. However, this active pulley hypothesis may simplify certain aspects of movements.12 For example, the pulleys may govern rotations of the eyes during saccades and pursuit so that they obey Listing’s law.14 Listing’s law states that when the eye rotates to a tertiary position, this is achieved by rotation around a single axis perpendicular to the primary position of gaze and to the new position of gaze. Together these axes form Listing’s plane, which is approximately frontoparallel. Donder’s law states that every tertiary eye position is associated with a specific torsional rotation. The noncommutative properties of rotations pose a computational challenge to the brain (the order of rotations must be specified), but the pulleys may simplify that problem mechanically in the orbit. It also seems possible that certain forms of congenital strabismus may be caused by misplacement of pulleys in the coronal plane.12 Precise measurements of eye rotations in all three directions,15 and high-definition imaging of the extraocular muscles are likely to clarify the role played by the extraocular muscle pulleys further.
TABLE 2. Actions Of The Extraocular Muscles With The Eye In Primary Position* | ||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||
PROPERTIES OF THE EXTRAOCULAR MUSCLES
Extraocular muscles differ anatomically and physiologically from limb muscles.16,17 The former have fibers that are smaller and more richly innervated; some extraocular muscle fibers are among the fastest contracting and yet are relatively fatigue-resistant. The extraocular muscles contain twitch fibers that have a single endplate per fiber and generate action potentials. In addition, there are nontwitch fibers that cannot generate action potentials but show graded contractions to trains of electrical pulse stimuli. These tonic fibers are capable of a smoothly modulated muscle contraction, which may be important for maintaining steady gaze. The extraocular muscles are selectively vulnerable to some disorders (e.g., myasthenia gravis) but resistant to others (e.g., Duchenne’s dystrophy). Furthermore, the appearances of diseases that affect the muscle are different when they involve the extraocular muscles. Both central global and peripheral orbital muscle layers contain fibers more suited for either sustained contraction or brief rapid contraction. However, the orbital layer contains many fatigue-resistant twitch fibers and the global zone contains twitch fibers with variable degrees of fatigue resistance; the different muscle types receive different innervation.13
Six types of fibers have been defined in the extraocular muscles.16,17 (i) In the orbital layer, approximately 80% of fibers are singly-innervated, have fast-type myofibrillar adenosine triphosphatase (ATPase), and high oxidative activity (with numerous mitochondria in dense clusters). They are very fatigue-resistant. (ii) The remaining orbital fibers are multiply innervated fibers, with multiple nerve terminals. They have twitch capacity near the center of the fiber, and nontwitch activity proximal and distal to end plate band. (iii) In the global layer, approximately 33% of fibers are red, singly innervated, fast-twitch, and fatigue-resistant. (iv) The global layer contains intermediate, singly innervated fibers (approximately 25%) with fast-twitch properties, numerous mitochondria, and intermediate level of fatigue resistance. (v) Global, pale singly innervated fibers (approximately 33%) have fast-twitch properties but contribute only sporadically because of their low fatigue resistance. (vi) Global multiply innervated fibers (approximately 10%) have slow-twitch, slow tonic properties that exhibit slow, graded, nonpropagated responses to neural or pharmacologic activation; this fiber type is unparalleled in any other human skeletal muscle. It should be noted that levator palpebrae lacks multiply innervated fibers; this may reflect the importance of this fiber type for fixation and smooth eye movements. Unlike other skeletal muscles, in which embryonic myosin is transformed to adult isoforms, extraocular muscles preserve their embryonic myosin in the proximal and distal portions of both types of orbital layer fibers.16 This may underlie the remarkable capacity of extraocular muscles to adapt to changes in innervation and disease states.
Ocular motoneurons and muscle fibers are the final common pathway for all ocular motor systems. The contribution that different fiber types make to different types of eye movements was clarified in classic experiments by Scott and Collins,18 who used miniature electrode needles with multiple recording sites. They reported that fibers are differentiated functionally by the amount of work performed and that the electromyographic (EMG) activity of a given unit correlates with an eye position, irrespectively of the type of movement. (It should be noted, however, that fibers that do not generate action potentials will not be evident on EMG). Orbital layer fibers are recruited before the global, but both types of fibers participate in every movement. Scott and Collins proposed a division of labor such that orbital fibers are active throughout nearly the entire range of movement but during fixation, global fibers are recruited only as the eye is called into the field of action of that muscle. The classic scheme of Scott and Collins is being reinterpreted in the light of the discovery of pulleys for the extraocular muscles and proprioceptive innervation of the extraocular muscles (discussed below).12,13
The different structure of the extraocular muscles apparently determines their differential involvement in neuropathic and myopathic pathologic processes. One example is myasthenia gravis (MG), an autoimmune condition with antibodies against acetylcholine receptors, in which extraocular muscles are preferentially affected.16,19 This may be accounted for by the difference in structure of the acetylcholine receptor in the extraocular muscles, in which the embryonic 2β type is preserved at the neuromuscular junction of multiply innervated fibers (in contrast to the adult 2β type of the receptor in the skeletal muscles).16,20 However, because multiply innervated fibers are absent from the levators, this explanation cannot account for frequent ptosis in myasthenic patients.21 Generally normal saccadic metrics in the presence of the affected ductions again argue for the preferential involvement of the multiply-innervated fibers, with preservation of the fast-twitch singly innervated fibers. Recently, it has been shown that injection of a standard dose of edrophonium (Tensilon) increased peak velocity-amplitude relationship in patients with MG while decreasing it in normal controls or patients with ocular palsies of other causes.22 Because clinical improvement in the amplitude is not specific for MG, the difference in the velocity response may prove to be of higher diagnostic value. The effect in normal controls may be the result of subclinical cholinergic excess (depolarizing blockade-like) and suggests that 10 mg is too high a dose.
Duchenne’s Muscular Dystrophy
In this systemic myopathy, as in most others, eye movements are spared.23 This correlates with the absence of necrosis in the extraocular muscles despite the deficiency of a subsarcolemmal protein, dystrophin.24 In other muscles, the initial pathology is increase in intracellular free calcium because of disruption of sarcolemmal integrity.16 It has been suggested that higher capacity of extraocular muscles to scavenge free radicals caused by higher levels of superoxide dismutase might account for this selective preservation of function.25
Although it has long been known that the extraocular muscles contain end-organs necessary for proprioception, their role in the control of eye movements has remained moot until recently.13 In part this has been because vision exerts much more powerful and immediate feedback control on eye movements than any non-visual signal. In addition, experimental studies in monkey have supported a role for the other extraretinal signal – efference copy or corollary discharge of eye movement commands;26 this internal neural signal is consistent with Helmholtz’s idea that the brain monitors its own effort of will. One conceptual reason to doubt that proprioception has any role in the control of eye movements is that no external loads are applied to the extraocular muscles, and evidence exists to refute the presence of a stretch reflex in the extraocular muscles.27
In extraocular muscles, the main sources of proprioceptive input are muscle spindles, which lie mainly in the orbital layer; myotendinous cylinders (palisade endings), which are associated with the global layer; and Golgi tendon organs that lie in the peripheral layer.28 It has been hypothesized that only twitch fibers play a substantial role in eye movement, whereas the global layer nontwitch muscle fibers adjust the tension on the palisade endings and modulate the afferent proprioceptive signal.28 Extraocular proprioceptors project to the brain via the ophthalmic branch of the trigeminal nerve and the Gasserian ganglion, to the spinal trigeminal nucleus (pars interpolaris and pars caudalis).29 From the trigeminal nucleus, proprioceptive information is distributed widely to structures involved in oculomotor control: superior colliculus, vestibular nuclei, nucleus prepositus hypoglossi, cerebellum, frontal eye fields, and also structures involved in visual processing: lateral geniculate body, pulvinar, visual areas 17 and 18.30 It has been shown that extraocular proprioception contributes to the development of visual binocularity, aspects of pattern recognition, and formation of visuospatial maps.30
Studies have suggested that ocular proprioception is important for adaptive recalibration, such as occurs after ocular motor palsies. Thus, after experimentally induced, unilateral extraocular palsies, it was shown that proprioceptive deafferentation of the paretic eye produced gradual worsening of both static alignment and saccadic conjugacy.31 Taken together, this evidence argues that proprioception contributes to long-term adaptive mechanisms responsible for eye alignment during fixation and saccades.
ANATOMY OF THE CRANIAL NERVES
The ocular motor nuclei are located in the brainstem, close to the midline. The intracranial courses of the ocular motor nerves are summarized in Figure 1.
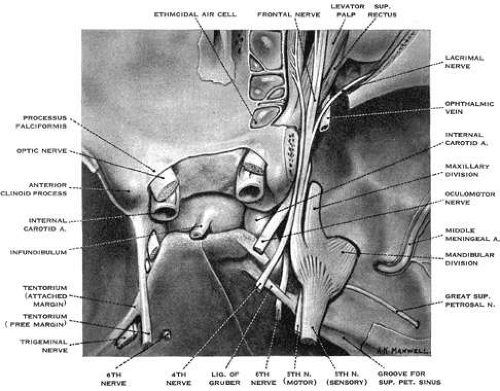 Fig. 1. The intracranial courses of the third, fourth, and sixth cranial nerves. Superior view. Lig. of Gruber: petroclinoid ligament. (From Warwick R. Wolff’s Anatomy of the Eye and Orbit, 7th ed. Philadelphia: WB Saunders, 1976:295, with permission) |
The abducens nucleus lies in the floor of the fourth ventricle, in the lower pons; it is capped by the genu of the facial nerve. As discussed below, the abducens nucleus contains two distinct populations of cells: motor neurons that innervate the lateral rectus muscle and internuclear neurons that innervate, via the medial longitudinal fasciculus, contralateral medial rectus motoneurons. From the medial aspect of the nucleus, fibers destined for the ipsilateral, lateral rectus muscle pass ventrally, laterally and caudally through the pontine tegmentum and medial lemniscus, and lateral to the corticospinal tract, to emerge from the pons at its caudal border. The abducens nerve then courses nearly vertically along the clivus, through the prepontine cistern, and rises to the petrous crest, where it bends forward to penetrate the dura. Here it lies medial to the trigeminal nerve, and passes under the petroclinoid ligament in Dorello’s canal. Because the nerve is relatively tethered in the last two locations, it is vulnerable to shear forces; thus, sixth nerve palsy is a nonlocalizing sign in the context of elevated intracranial pressure or trauma. In the cavernous sinus, it lies lateral to the internal carotid artery and medial to the ophthalmic division of the trigeminal nerve. For a short portion, pupillosympathetic fibers run with the sixth nerve as they leave the carotid artery to reach the first division of the trigeminal nerve. Therefore, the combination of the sixth-nerve palsy with Horner’s syndrome is suggestive of a cavernous sinus process. The sixth nerve then enters the orbit through the superior orbital fissure, and passes through the annulus of Zinn to innervate the lateral rectus on its inner surface. The most common causes of the sixth-nerve palsy are (peripheral) vascular disease caused by diabetes mellitus or hypertension in an elderly population, and tumors in younger patients.
The trochlear nucleus lies at the ventral border of the periaqueductal gray matter at the level of the inferior colliculus. It innervates the contralateral superior oblique muscle. The trochlear nerve is the longest and the thinnest of cranial nerves; therefore, it is susceptible to even minor head trauma. Its fibers pass dorsolaterally and caudally, around the central gray matter, and decussate completely in the roof of the aqueduct, within the superior medullary velum. Lesions there can produce bilateral fourth nerve palsies. The trochlear nerve emerges from the dorsal aspect of the brainstem, caudal to the inferior colliculus, and close to the tentorium cerebelli, and passes laterally around the upper pons to reach the prepontine cistern. It then runs forward on the free edge of the tentorium before entering the cavernous sinus. Within the lateral wall of the sinus the fourth nerve lies below the third nerve and above the ophthalmic division of the fifth nerve. It crosses over the oculomotor nerve to enter the superior orbital fissure and passes to the medial aspect of the orbit to supply the superior oblique muscle.
The oculomotor nucleus is a paramedian structure that lies at the ventral border of the periaqueductal gray matter. It extends rostrally to the level of the posterior commissure and caudally to the trochlear nucleus. It sends fibers to the medial rectus, superior rectus, inferior rectus and inferior oblique muscles, and to the levator palpebrae superioris. Warwick’s anatomic scheme for the oculomotor nucleus32 has been revised with the demonstration that the neurons supplying the medial rectus muscle are distributed into three areas of the oculomotor nucleus.33 The neurons innervating each superior rectus muscle lie next to each other, and their axons decussate in this part of the nucleus. The central caudal nucleus, which supplies both levator palpebrae superioris muscles, is a single structure. All projections from the oculomotor nucleus are ipsilateral except for those to the superior rectus, which are totally crossed, and those to the levator palpebrae superioris, which are bilateral.
The fascicles of the oculomotor nerve pass ventrally through the red nucleus, the substantia nigra, and the medial part of the cerebral peduncle. A topographic organization has been proposed on the basis of the effects of clinical lesions; from lateral to medial, the order is inferior oblique, superior rectus, medial rectus and levator palpebrae, inferior rectus, and parasympathetic pupillary fibers from Edinger-Westphal nucleus.34 The rootlets of the third nerve emerge from the interpeduncular fossa and then fuse and pass between the posterior cerebral artery and superior cerebellar artery into the basal cistern. The third nerve passes lateral to the posterior communicating artery and below the temporal lobe uncus, where it runs over the petroclinoid ligament just lateral to the posterior clinoid process. During its subarachnoid course, parasympathetic pupillary fibers lie in the peripheral, dorsomedial part of the nerve. Their peripheral location, however, is not the only reason for the pupillary involvement with structural lesions of the third nerve; early pupillary involvement also reflects pressure-sensitive nature of these fibers. The oculomotor nerve pierces the dura close to the free edge of the tentorium cerebelli. Within the cavernous sinus, the third nerve lies initially above the trochlear nerve, where it receives sympathetic fibers from the carotid artery. As it leaves the cavernous sinus, it divides into superior and inferior branches; these pass through the superior orbital fissure. The superior branch runs laterally to the optic nerve and ophthalmic artery to supply the superior rectus and levator palpebrae muscles. The inferior branch supplies the medial rectus, inferior rectus and inferior oblique muscles, and the ciliary ganglion. The most common cause of isolated third nerve palsy is vascular disease (in association with diabetes or hypertension); in such cases the pupil is usually spared. The second most common cause are aneurysms, typically of the supraclinoid portion of the internal carotid or posterior communicating artery; such cases characteristically have pupillary involvement; exceptions to this rule, however, have been reported.
BRAINSTEM CONTROL OF HORIZONTAL GAZE
MOTOR COMMANDS FOR CONJUGATE HORIZONTAL MOVEMENTS
The abducens nucleus is of prime importance in the control of horizontal gaze because it governs conjugate movements of both the ipsilateral lateral rectus and the contralateral medial rectus muscles. It contains two populations of neurons: (i) abducens motoneurons, which supply the lateral rectus muscle, and (ii) abducens internuclear neurons, which project up the contralateral medial longitudinal fasciculus (MLF) to synapse on medial rectus motoneurons of the oculomotor nucleus (Fig. 2).35,36 Thus, axons of the abducens nerve, and axons of the abducens internuclear neurons that run in the MLF,37 together encode the conjugate, horizontal eye movement command. In addition, oculomotor internuclear neurons, which lie in the medial rectus subdivision of the oculomotor nucleus, project to the contralateral abducens.38 Together, these two types of internuclear neurons are responsible for yoking of conjugate horizontal movements known as Hering’s law. Both abducens motoneurons and abducens internuclear neurons receive similar afferent input from each functional class of eye movement (Table 1).39.40 The abducens nucleus receives excitatory vestibular and optokinetic afferents from the contralateral vestibular nuclei.41,42 Saccadic commands originate from burst neurons of the pontine and medullary reticular formation.43,44 A descending smooth pursuit pathway projects to the abducens nucleus via the vestibular and cerebellar fastigial nuclei (discussed further below). The signals that are necessary for eccentric gaze-holding function reach the abducens nucleus from the nucleus prepositus hypoglossi and the medial vestibular nuclei.45
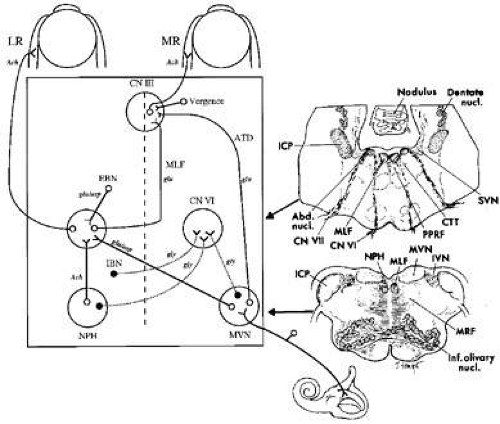 Fig. 2. Anatomic scheme for the synthesis of signals for horizontal eye movements. The abducens nucleus (CN VI) contains abducens motoneurons that innervate the ipsilateral lateral rectus muscle (LR), and abducens internuclear neurons that send an ascending projection in the contralateral medial longitudinal fasciculus (MLF) to contact medial rectus (MR) motoneurons in the contralateral third nerve nucleus (CN III). From the horizontal semicircular canal, primary afferents on the vestibular nerve project mainly to the medial vestibular nucleus (MVN), where they synapse and then send an excitatory connection to the contralateral abducens nucleus and an inhibitory projection to the ipsilateral abducens nucleus. Saccadic inputs reach the abducens nucleus from ipsilateral excitatory burst neurons (EBN) and contralateral inhibitory burst neurons (IBN). Eye position information (the output of the neural integrator) reaches the abducens nucleus from neurons within the nucleus prepositus hypoglossi (NPH) and adjacent MVN. The medial rectus motoneurons in CN III also receive a command for vergence eye movements. Putative neurotransmitters for each pathway are shown: Ach, acetylcholine; asp, aspartate; glu, glutamate; gly, glycine. The anatomic sections on the right correspond to the level of the arrow heads on the schematic on the left. Abd. nucl., abducens nucleus; CN VI, abducens nerve; CN VII, facial nerve; CTT, central tegmental tract; ICP, inferior cerebellar peduncle; IVN, inferior vestibular nucleus; Inf. olivary nucl., inferior olivary nucleus; MVN, medial vestibular nucleus; MRF, medullary reticular formation; SVN, superior vestibular nucleus. (Modified from Leigh RJ, Zee DS. The Neurology of Eye Movements, 3rd ed. New York: Oxford University Press, 1999) |
EFFECTS OF LESIONS OF THE PONS AND MEDULLA ON HORIZONTAL GAZE
Lesions of the abducens nucleus cause paralysis of both the ipsilateral lateral rectus and contralateral medial rectus muscles for all conjugate eye movements,46,47,48 but vergence movements are spared. Clinical lesions restricted to the abducens nucleus are rare, and often there is also involvement of adjacent structures including the facial nerve fascicle, MLF, and paramedian pontine reticular formation (PPRF).
Lesions of the medial longitudinal fasciculus produce internuclear ophthalmoplegia (INO), which is characterized by paresis of adduction, for conjugate movements, on the side of the lesion49,50; in addition there is usually dissociated nystagmus, characterized by abduction overshoot of the eye contralateral to the lesion. This nystagmus is not an inherent feature of the MLF lesion but probably reflects efforts of central adaptive mechanisms to correct the adduction weakness.51 Thus, dissociated nystagmus is also described with other disorders that cause selective medial rectus weakness. INO is often accompanied by skew deviation, caused by disruption of otolithic-ocular projections that run in the MLF.52 In addition, bilateral INO is usually associated with gaze-evoked vertical nystagmus, impaired vertical pursuit, and decreased vertical vestibular responses.53 Small-amplitude saccadic intrusions may interrupt fixation.54 This could be attributed to the associated lesion of the adjacent omnipause neurons or their connections.55 The most frequent cause of INO in young adults, especially when bilateral, is multiple sclerosis; other causes are summarized in Table 3.
TABLE 3. Causes of Internuclear Ophthalmoplegia* | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
When INO is subtle, it is best identified by comparing the speed and conjugacy of a series of large horizontal saccades, looking for relative slowing of the adducting movement. A series of quick phases of nystagmus, induced with an optokinetic tape, is another way to compare relative speed of adducting and abducting movements. Some care is required in interpreting these signs, however, because abducting saccades are faster in normal subjects, and quantitative comparison of the velocity of the two eyes may be required to make the diagnosis in subtle cases.56,57
Patients with INO may be orthotropic (or exophoric) in primary position without symptomatic diplopia unless accompanied by skew deviation. Some patients with bilateral INO show exotropia (wall-eyed bilateral INO syndrome). The explanation for exotropia is unclear, however, because monkeys with a lidocaine-induced internuclear ophthalmoplegia show an increased accommodative vergence to accommodation ratio, implying that the MLF actually carries signals that inhibit vergence.58 Furthermore, exotropia may occur in patients with INO who have preserved convergence.1 The classification of INO into anterior and posterior types depending on the integrity of the convergence mechanism is not of localizing value in identifying the rostral-caudal location of the MLF lesion. The so-called posterior internuclear ophthalmoplegia of Lutz,59 in which abduction (but not adduction) is impaired is rare. It has been difficult to account for because the abducens contains neurons that innervate both lateral and medial rectus muscles for all conjugate eye movements;60 one possible explanation may be dysfunction of the descending excitatory projections from the third to the sixth nucleus.38
A combined lesion of one MLF and the adjacent abducens nucleus (or its inputs) produces paralysis of all conjugate movements save for abduction of the eye contralateral to the side of the lesion: one-and-a-half syndrome.61,62 When the lesion is acute, the patient may be exotropic63; the deviated eye may show nystagmus and is on the side opposite that of the brainstem lesion. This syndrome occurs with a variety of causes including brainstem infarction, hemorrhage, multiple sclerosis, and pontine glioma.
Discrete lesions of the PPRF,64,65 which mainly corresponds to the nucleus pontis centralis caudalis and contains saccadic burst neurons, cause loss of horizontal saccades and quick phases of nystagmus to the side of the lesion. Bilateral, destructive PPRF lesions may also produce slow vertical saccades. Selective bilateral impairment of horizontal saccades occurs in degenerative conditions, such as variants of olivopontocerebellar atrophy.66 Other causes of slow or absent saccades are summarized in Table 4. Infarction of the paramedian pons may also involve axons conveying vestibular and pursuit inputs to the abducens nucleus.67 Pontine disease may cause a unilateral defect of smooth pursuit by affecting the dorsolateral pontine nuclei and their projections to the cerebellum (see below).68 More rostral brainstem lesions tend to cause ipsilateral smooth pursuit deficits, whereas caudal brainstem and cerebellar lesions cause contralateral deficits.69 Unilateral lesions of the vestibular nerve or labyrinth cause vertigo, nystagmus, and skew deviation (see below).1 Bilateral peripheral vestibular lesions cause impaired vision and oscillopsia during head movements, especially those occurring during locomotion.7 Lesions affecting the central vestibular nuclei also cause an imbalance that is manifest by nystagmus and skew deviation. When such lesions involve the medial vestibular nuclei (MVN) and adjacent nucleus prepositus hypoglossi (NPH), they also impair the ability to hold eccentric gaze and produce gaze-evoked nystagmus. Thus, bilateral, experimental lesions of MVN and NPH abolish the eccentric gaze-holding mechanism for eye movements in the horizontal plane.70 In health, a network of neurons in the brainstem MVN and NPH as well as the vestibular cerebellum generates the eye position command required to hold the eye steady in an eccentric orbital position. Following lesions of this neural network, the eye cannot be held in an eccentric position in the orbit and it drifts back to primary position. Corrective quick phases then produce gaze-evoked nystagmus.
In lateral medullary infarction (Wallenberg’s syndrome), there may be spontaneous nystagmus that is usually horizontal or mixed horizontal-torsional with the slow phases directed toward the side of the lesion; the nystagmus may reverse direction in eccentric positions, suggesting coexistent involvement of the gaze-holding mechanism. Skew deviation, with an ipsilateral hypotropia, cyclodeviation (lower eye more extorted), and ipsilateral head tilt (when present together called the ocular tilt reaction) reflect an imbalance of otolithic inputs (see below).71 In addition, patients with Wallenberg’s syndrome show a characteristic lateropulsion in which the trajectory of saccades, whether horizontal or vertical, is deviated towards the side of the lesion.72 A hypothetical explanation for lateropulsion is discussed further below.
BRAINSTEM CONTROL OF VERTICAL AND TORSIONAL GAZE
The oculomotor and trochlear nuclei contain the motoneurons for vertical and torsional eye movements. Thus, the substrate for each functional class of eye movements must project to these motoneurons (Fig. 3). On the one hand, vertical and torsional saccades and the mechanism for eccentric vertical gaze-holding are synthesized in the midbrain. On the other hand, vestibular and pursuit signals ascend to the midbrain from the lower brainstem.
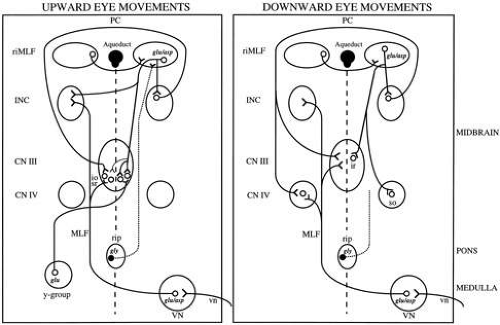 Fig. 3. Anatomic schemes for the synthesis of upward, downward, and torsional eye movements. From the vertical semicircular canals, primary afferents on the vestibular nerve (vn) synapse in the vestibular nuclei (VN) and ascend in the medial longitudinal fasciculus (MLF) and brachium conjunctivum (not shown) to contact neurons in the trochlear nucleus (CN IV), oculomotor nucleus (CN III), and the interstitial nucleus of Cajal (INC). The rostral interstitial nucleus of the medial longitudinal fasciculus (riMLF), which lies in the prerubral fields, contains saccadic burst neurons. It receives an inhibitory input from omnipause neurons of the nucleus raphe interpositus (rip), which lie in the pons (for clarity, this projection is only shown for upward movements). Excitatory burst neurons in riMLF project to the motoneurons of CN III and CN IV, and also send an axon collateral to INC. Each riMLF neuron sends axon collaterals to yoke-pair muscles (Hering’s law). Projections to the elevator subnuclei (innervating the superior rectus and inferior oblique muscles) may be bilateral due to axon collaterals crossing at the level of the CN III nucleus. Projections of inhibitory burst neurons are less well understood, and are not shown. The INC provides a gaze-holding signal, and projects to vertical motoneurons via the posterior commissure. Signals contributing to vertical smooth pursuit and eye–head tracking reach CN III from the y-group via the brachium conjunctivum and a crossing ventral tegmental tract. Neurotransmitters: asp, aspartate; glu, glutamate; gly, glycine. (Modified from Leigh RJ, Zee DS. The Neurology of Eye Movements, 3rd ed. New York: Oxford University Press, 1999) |
In the prerubral fields at the junction of midbrain and diencephalon, lies the rostral interstitial nucleus of the medial longitudinal fasciculus (riMLF),73 which has also been called the nucleus of the prerubral fields and the nucleus of the fields of Forel. It contains burst neurons for vertical saccades and quick phases, and for torsional quick phases. Each riMLF contains neurons that burst for both upward and downward eye movements, but for torsional quick phases in only one direction. For example, the right riMLF discharges for quick phases that are directed clockwise with respect to the subject.74 Each riMLF is connected to its counterpart commissural projections. The riMLF projects to motoneurons innervating elevator muscles bilaterally, but to motoneurons innervating depressor muscles ipsilaterally; therefore each riMLF is responsible for innervation of the contralateral superior oblique and ipsilateral inferior rectus muscles for saccades.75,76 Furthermore, each burst neuron in the riMLF sends axon collaterals to motoneurons supplying yoke muscle pairs; this appears to be part of the neural substrate for Hering’s law in the vertical plane.77
The riMLF also projects to the interstitial nucleus of Cajal (INC), which has been shown to play an important role in vertical gaze-holding.78 In addition, the INC also receives inputs from the vestibular nuclei.79 The INC projects via the posterior commissure to motoneurons of the contralateral vertical ocular motor subnuclei.80 The INC also contains neurons that project to motoneurons of the neck and trunk muscles, and appears to coordinate combined torsional-vertical movements of the eyes and head. The neural signals necessary for vertical vestibular and smooth pursuit eye movements ascend to INC from the medulla and pons. The MLF is the most important route for these projections but the brachium conjunctivum and other pathways are also involved.81
EFFECTS OF MIDBRAIN LESIONS ON VERTICAL AND TORSIONAL GAZE
Experimental unilateral lesions of the riMLF cause a mild defect in vertical saccades, because nucleus on each side contains burst neurons for both upward and downward movements. A right riMLF lesion would also have little effect on upward saccades but downward saccades might be slowed82,83; this asymmetry of vertical defects may reflect the bilateral projections of riMLF to elevator muscles but ipsilateral projections to depressor muscles. However, a unilateral riMLF lesion produces a specific defect of torsional quick phases.82,83 For example, with a lesion of the right riMLF, torsional quick phases, clockwise from the point of view of the subject (extorsion of the right eye and intorsion of the left eye) are impaired.
Vertical saccadic deficits with unilateral lesions of the riMLF in humans are rare and often may reflect involvement of the commissural pathways of the riMLF that makes the lesion, in effect, bilateral.82,84 Thus, one reported patient with a unilateral midbrain lesion involving riMLF, but no evidence of commissural involvement, manifested contralesional tonic ocular tilt reaction (see below), torsional nystagmus and impaired vertical gaze, especially for downward saccades that became both limited and slow.85 In general, however, bilateral lesions are required to produce clinically apparent deficits of vertical eye movements.86 Bilateral experimental lesions of the riMLF in monkeys cause a defect of vertical and torsional saccades that may be more pronounced for downward eye movements83,87; vertical gaze-holding, vestibular eye movements and, possibly, pursuit are preserved, as are horizontal saccades. Patients with discrete, bilateral infarction in the region of the riMLF show deficits of either downward or both upward and downward saccades.88 Extensive bilateral PPRF lesions cause slow vertical saccades by impairing the ascending input to the riMLF. Certain metabolic and degenerative disorders may lead to selective slowing or absence of vertical saccades (Table 4).
TABLE 4. Causes Of Slow Saccades* | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
Unilateral experimental lesions of the INC are reported to impair gaze-holding function in the vertical plane and reduce the vertical ocular motor range.78 In addition, skew deviation (ipsilateral hypertropia), extorsion of the contralateral eye, intorsion of the ipsilateral eye, and contralateral head tilt occurs. Stimulation near the INC in the monkey produces an ocular tilt reaction that consists of an ipsilateral head tilt and a synkinetic ocular reaction: depression and extorsion of the eye ipsilateral to the stimulation and elevation and intorsion of the contralateral eye.89 This ocular tilt reaction is similar to that produced by stimulation of the contralateral utricular nerve or an ipsilateral lesion of the vestibular nucleus (see Wallenberg’s syndrome, above). Bilateral lesions of the medial longitudinal fasciculus (bilateral INO) impair vertical vestibular and pursuit movements but spare vertical saccades.90 In addition, partial loss of the vertical eye position signal causes vertical gaze-evoked nystagmus.
Lesions of riMLF and INC are both often associated with torsional nystagmus. While tonic torsional deviation in both cases is contralesional, the direction of the fast phases for the nystagmus varies: with riMLF lesions, the nystagmus is contralesional whereas with INC lesions, it is ipsilesional.85
Lesions of the posterior commissure are equivalent to bilateral lesions of the INC, and reduce the range of vertical movements, especially upward (Parinaud’s syndrome)91; usually all types of eye movement are affected, although the vestibulo-ocular reflex and Bell’s phenomenon may sometimes be spared. Experimental inactivation of the posterior commissure with lidocaine impairs vertical gaze-holding function.92 In addition, posterior commissure lesions may cause slowing of vertical saccades below the horizontal meridian, and disorders of convergence including convergence-retraction nystagmus. Attempts at upward saccades evoke convergence-retraction nystagmus that persists as long as the refixation effort is maintained. This is best elicited with a down-moving optokinetic tape stimulating a series of repetitive upward saccades; a convergence or retraction movement replaces each fast phase. These movements may or may not truly be nystagmus but consist of opposed adducting saccades followed by slow divergence movements.93,94 With dorsal midbrain lesions, abduction may be bilaterally impaired, perhaps reflecting increased vergence gain, giving the appearance of pseudoabducens palsy.95 Rarely, a divergence-retraction nystagmus may occur in patients with dorsal midbrain syndrome. Other associated findings with pretectal lesions are pathologic lid retraction (Collier’s sign), and middilated pupils that show a smaller reaction to light than to the near stimulus. Pineal area tumors96,97 and midbrain infarction84,88 are the most common causes.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


