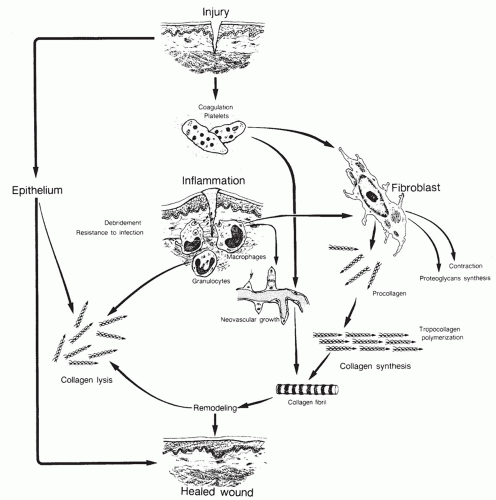
 Figure 6.1 Brief depiction of the complexity of wound healing. (Modified and reprinted from Feinberg SE, Larson PE. Healing of traumatic injuries. In: Fonseca RJ, Walker RV, eds. Oral and maxillofacial trauma. Philadelphia, PA: WB Saunders, 1991, with permission.) |
TABLE 6.1 CYTOKINES INVOLVED IN WOUND HEALING | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
chemotaxis of neutrophils, macrophages, endothelial cells, and fibroblasts. Platelets also contain vasoactive amines like serotonin that cause vasodilation and increased vascular permeability. Fluid extravasation occurs contributing the edema observed during the second phase of wound healing, the inflammatory phase (2,3).
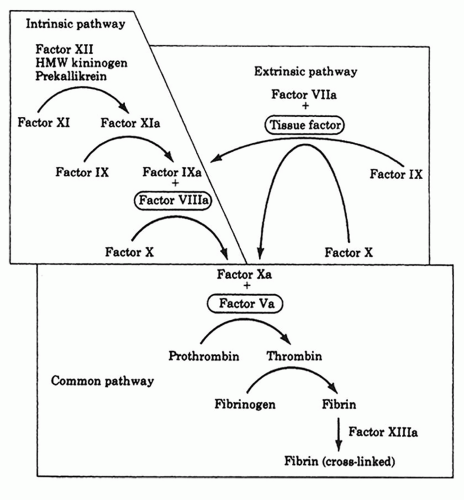 Figure 6.2 The coagulation cascade. (From Klingensmith ME. Washington manual of surgery, 5th ed. Lippincott Williams & Wilkins, 2005, with permission.) |
and collagenase. Activated keratinocytes, fibroblasts, and endothelial cells also play a role (4).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


