Chapter 89 Drug Toxicity of the Posterior Segment
A variety of systemic medications can generate retinal toxicity. Fortunately, in the majority of cases the loss of visual function is minimal or reversible following discontinuation of the inciting drug. Nevertheless, permanent or progressive visual loss may occur in some instances. We present those medications known to produce a well-described anomaly and have omitted others that have not been definitively proven to cause retinal abnormalities. The medications are grouped according to the type of retinal toxicity they produce, summarized in Box 89.1.
Box 89.1
Patterns of retinal toxicity
Disruption of the retina and retinal pigment epithelium
Phenothiazines
Thioridazine
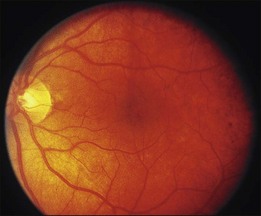
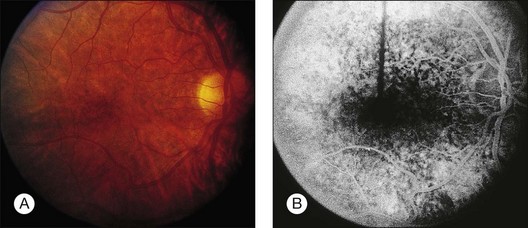
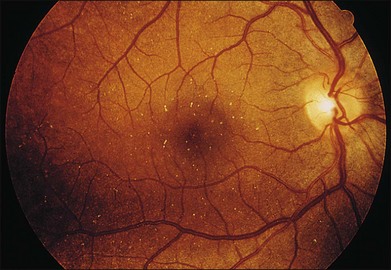
Blurred vision, dyschromatopsia (reddish or brownish discoloration of vision), and nyctalopia characterize acute toxicity with thioridazine.1 In the earliest stages the fundus appearance may be normal or display only mild granular pigment stippling (Fig. 89.1). An intermediate stage is characterized by circumscribed nummular areas of retinal pigment epithelial (RPE) loss from the posterior pole to the midperiphery2 (Fig. 89.2A). Fluorescein angiography (FA) reveals disruption of the choriocapillaris in these zones of pigment rarefaction (Fig. 89.2B). In late stages of thioridazine toxicity, widespread areas of depigmentation alternating with hyperpigmented plaques, vascular attenuation, and optic atrophy are seen3 (Fig. 89.3).
Retinal toxicity from thioridazine is dependent more on the total daily dose than on the cumulative amount of drug received.4 With higher daily doses, toxicity can occur rapidly, even within the first two weeks of therapy.5 Toxicity is rare at dosages less than 800 mg/day. Nonetheless, a few cases have been reported with lower doses given over several years.6–10 As a result, many now suggest that any patient taking thioridazine, regardless of the daily dose, should be monitored for the development of visual symptoms or fundus changes.
In the initial stages of toxicity, visual field testing can reveal mild constriction, paracentral scotomas, or ring scotomas. Electroretinography (ERG) is either normal or shows decreased oscillatory potentials. In the later stages, both the rod and cone functions of the ERG, as well as electrooculography (EOG), are markedly abnormal.11 If the drug is stopped early, ERG testing often improves over the first year.12 Histologic studies demonstrate that atrophy and disorganization of photoreceptor outer segments occurs primarily, with a secondary loss of the RPE and choriocapillaris.3
The early fundus changes associated with thioridazine often progress despite discontinuation of therapy.2 It is unclear whether this degeneration represents continued toxicity of the drug or a delayed expansion of chorioretinal scarring to areas of subclinical, preexisting damage.12 Visual function, in contrast to fundus appearance, usually improves over the first year after a toxic reaction, but there has been one report of severe progressive decline in vision after cessation of the drug.13
The mechanism of thioridazine-mediated toxicity remains unknown. Many phenothiazines bind melanin granules of the RPE and uveal tissue, but not all commonly instigate retinal toxicity.14–16 The compound NP-207 (piperidyl-chlorophenothiazine hydrochloride) has a remarkably similar chemical structure to thioridazine, including the same piperidyl side chain. NP-207 was never marketed because of the pronounced pigmentary retinopathy that developed during early clinical trials.17 This piperidyl side chain is not present in other phenothiazines such as chlorpromazine, which exhibit much less retinal toxicity. Experimental studies demonstrate that phenothiazines both alter enzyme kinetics and inhibit oxidative phosphorylation with subsequent abnormalities in rhodopsin synthesis.18–20 Other studies postulate that phenothiazine toxicity is due to the drug’s effect on the dopamine receptors in the retina.21 Further study is necessary to determine whether these observed effects are involved in the pathogenesis of thioridazine toxicity.
Chlorpromazine
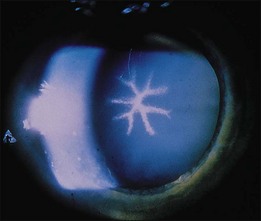
Chlorpromazine is a piperazine similar to thioridazine but lacks the piperidyl side chain mentioned above. The compound binds strongly to melanin and can cause hyperpigmentation in the skin, conjunctiva, cornea, lens, and retina22–28 (Fig. 89.4). Other ocular effects include oculogyric crisis, miosis, and blurred vision caused by paralysis of accommodation. Usual doses range from 40 to 75 mg/day, but dosages up to 800 mg/day are not uncommon.
Retinal toxicity from chlorpromazine is rare. When massive doses are given (e.g., 2400 mg/day for 12 months), pigmentary changes may occur in the retina with attenuation of retinal vessels and optic nerve pallor25 (Fig. 89.5). Similar to thioridazine, the development and extent of toxicity are more closely related to daily dosage than total amount of drug taken.
Chloroquine derivatives
Chloroquine
Chloroquine was first used as an antimalarial drug in World War II. Currently it is prescribed for treatment of amebiasis, rheumatoid arthritis, and systemic lupus erythematosus in countries primarily outside the United States, and for prophylaxis against malaria. Retinal toxicity with degeneration of the RPE and neurosensory retina as a result of long-term daily use of chloroquine has been well described.29–35 However, most cases of retinopathy have developed when a higher than currently recommended (3 mg/kg/day using lean body weight) dose was used.36A daily dose exceeding 250 mg with a total cumulative dose between 100 and 300 g is customarily needed to produce toxicity.37 One study showed a 19% incidence of chloroquine retinopathy in patients taking a mean daily dose of 329 mg.38 Conversely, with strict adherence to a low dose per diem, the incidence of retinal abnormalities is minimal even when cumulative doses reach over 1000 g.39
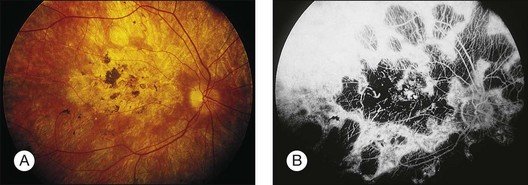
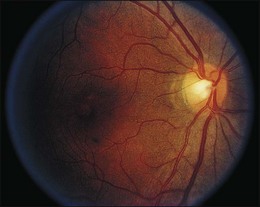
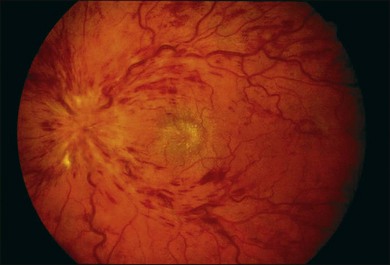
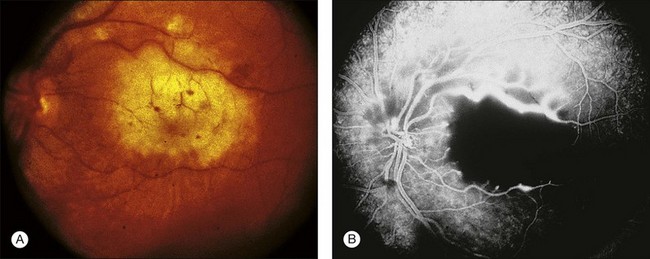
A paracentral scotoma may be the earliest manifestation of retinal toxicity and can precede the development of any ophthalmoscopic or ERG abnormality.40 Subtle macular pigment stippling with a loss of the foveal light reflex (Fig. 89.6) usually appears on fundus examination before the development of a classic bull’s-eye maculopathy, in which a ring of depigmentation surrounded by an area of hyperpigmentation is seen centered on the fovea (Fig. 89.7). Visual acuity decreases when the RPE abnormalities involve the center of the fovea. The peripheral retina can display pigment mottling, which may, in severe cases, develop into the appearance of primary tapetoretinal degeneration with narrowed retinal vessels, optic disc pallor, and eventual blindness (Fig. 89.8).
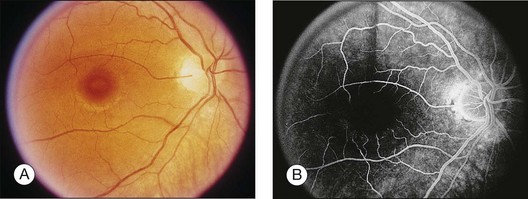
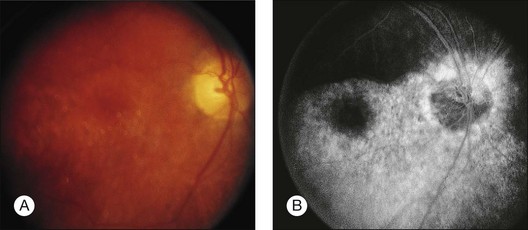
Fig. 89.7 Advanced chloroquine toxicity. Later photograph (A) and fluorescein angiogram (B) from the patient in Fig. 89.6 show marked progression with advanced widespread pigmentary changes.
(Reproduced with permission from Mieler WF. Focal points. American Academy of Ophthalmology, December 1997.)
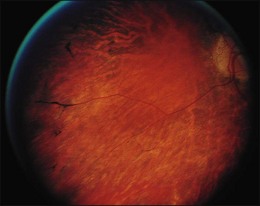
After the cessation of chloroquine treatment, early subtle macular changes can revert to normal. Although far advanced cases may progress despite discontinuation of the drug, most patients remain stable with long-term follow-up.41,42 Chloroquine, however, is very slowly excreted from the body. It has been detected in the plasma, red blood cells, and urine of patients 5 years after their last known ingestion.43 This prolonged presence may account for the rare cases of delayed onset of chloroquine retinopathy seen up to 7 years or longer after discontinuation.44,45
Fluorescein angiography can be helpful in the early demonstration of pigment abnormalities in the macula (see Figs 89.6, 89.7). There is minimal evidence of damage to the choriocapillaris on FA in the areas of pigment disturbance. The ERG and EOG may be abnormal early, although the EOG is sometimes supernormal initially and is not as helpful diagnostically.46 Histopathologic sections demonstrate loss of RPE pigmentation with an accumulation of pigment-laden cells in the outer retinal layers with damage and reduction of photoreceptors.47 Electron microscopic studies reveal more widespread damage to the retina, especially the ganglion cell layer.48 The retinal nerve fiber layer thickness has been shown to be significantly decreased compared to normal in patients on chloroquine therapy and is correlated to the daily dose taken.49 Fundus autofluorescence (FAF) and optical coherence tomography (OCT) findings suggest that the ganglion cell layer is affected by toxicity initially, especially surrounding the retinal vasculature.50
Like the phenothiazines, chloroquine is bound by melanin and concentrated in the RPE and uveal tissues.51 It appears that chloroquine toxicity may be mediated by disruption of lysosomal function in the RPE and neural retina and by inhibition of critical enzymes and interference with their metabolic function.44,52,53
With the availability of hydroxychloroquine, a less toxic but similar medication, use of chloroquine has steadily waned. Screening for toxicity has been more fully evaluated recently for hydroxychloroquine (see below), and similar testing is likely appropriate for chloroquine as well. Color vision can be abnormal in early toxicity and use of the Standard Pseudoisochromatic Plates Part 2 (SPP-2) or the American Optical Hardy Rand Rittler (AO-HRR) color vision plates provides adequate sensitivity and specificity for detection of abnormalities.54,55 Multifocal ERG testing may be abnormal in early toxicity even when other tests such as visual field and full-field ERG are normal.56,57
Hydroxychloroquine
Given the incidence of toxicity with chloroquine, most rheumatologists prefer hydroxychloroquine for the treatment of rheumatoid arthritis and systemic lupus erythematosus. Although it can produce a retinopathy identical to chloroquine, its occurrence is much less common.58–61 Toxicity involving decreased visual acuity, paracentral scotoma, and a bull’s-eye maculopathy has been documented62–67 (Figs 89.9, 89.10). Many of these patients received above the recommended daily dosage of 6.5 mg/kg/day, but the classic fundus findings have been reported at lower doses as well.62,64,67–69 Screening for toxicity becomes more important the longer the patient has been taking the drug as toxicity approaches 1% after 5–7 years of therapy and/or a cumulative dose of 1000 g.70–72
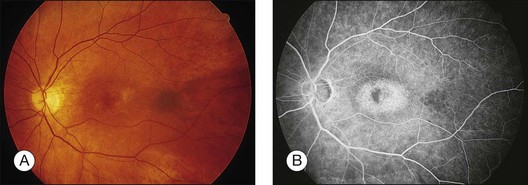
Several authors have questioned the utility of screening given the low yield, high cost, and the difficulty in diagnosing the condition early enough to prevent damage.73–76 Nevertheless, if retinal and functional changes are detected early, severe visual impairment can be averted.69,77
The revised American Academy of Ophthalmology guidelines for screening include a baseline examination performed at the commencement of therapy.78 Screening exams during the first five years of therapy can be performed during routine ophthalmic examination (interval to be determined by the age of the patient and the presence or absence of retinal or macular disease). Earlier recommendations emphasized dosing by weight. As most patients are given 400 mg/day of hydroxychloroquine, this dose is acceptable for all except for those with short stature (generally 5 feet 2 inches or less in height). These patients should be given a dose based on their ideal body weight, otherwise overdosage may occur.79 Furthermore, the dosage may need to be altered if the patient has renal or liver dysfunction.
After five years of therapy, screening should be performed at least annually.71 Current guidelines are centered around tests found to detect early toxicity often prior to any appreciable fundus findings. Patients should have a Humphrey 10–2 automated visual field test with a white test object and in addition should have one of three objective tests at each screening: multifocal electroretinogram (mfERG),70,80–84 spectral domain OCT,85–87 and/or FAF88 (Fig. 89.11). Any abnormalities of the pattern deviation on the Humphrey 10–2 visual fields should be taken seriously and the test repeated to confirm its reproducibility. In most situations, and since SD-OCT testing is so readily available, SD-OCT should also be obtained. While abnormalities on FAF are generally associated with concerns for active disease, the test has not yet been shown to be reliably predictable as a screening tool for future toxicity.
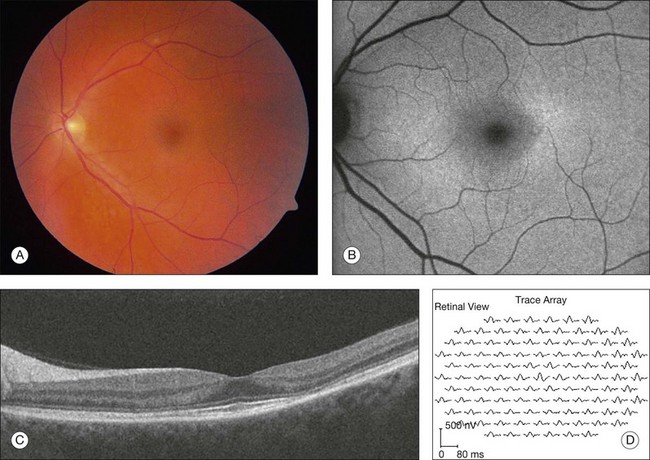
As noted in the preceding paragraph, it is imperative to discuss the risk of toxicity with patients and the rationale for screening (to detect, but not necessarily prevent visual loss).78 If ocular toxicity occurs,63,65,66,89 and is recognized at an early stage, efforts should be made to communicate this directly to the prescribing physician so that alternative treatment options can be discussed with the patient. In almost all cases, cessation of the drug should be suggested.
Quinine sulfate
Quinine sulfate was first used for the treatment of malaria in World War II, but it currently is prescribed for the management of nocturnal muscle cramps or “restless leg syndrome”. The recommended daily dose is less than 2 g. Signs of systemic toxicity occur with doses greater than 4 g, and the fatal oral dose is 8 g. Ocular toxicity with quinine develops after an overdose, either by accidental ingestion or by attempted abortion or suicide. Rarely, chronic ingestion at low levels can result in ocular toxicity as well.90 With an overdose, a syndrome known as cinchonism is rapidly produced, consisting of nausea, vomiting, headache, tremor, and sometimes hypotension and loss of consciousness. When patients awake they often are completely blind and have dilated, unreactive pupils.91 In the acute stages of toxicity, fundus examination reveals mild venous dilation with minimal retinal edema and normal arterial caliber. The FA displays minimal abnormalities. ERG testing shows an acute slowing of the a-wave with increased depth, loss of oscillatory potentials, and a decreased b-wave.92 EOG and visual-evoked potential (VEP) testing are also abnormal.
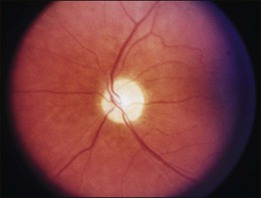
Over the next few days visual acuity returns, but the patient is left with a small central island of vision. There is a progressive attenuation of the retinal arterioles with the development of optic disc pallor over the next few weeks to months and iris depigmentation can occur93 (Fig. 89.12). Early investigators believed the mechanism of quinine toxicity to be vascular in origin. This was based primarily on the fundus appearance several weeks after ingestion, which showed marked arteriolar attenuation and optic disc pallor.91,94 More recent experimental and clinical studies have demonstrated minimal involvement of the retinal vasculature in the early stages of quinine toxicity.91,94,95 Furthermore, ERG and histologic studies show that the site of toxicity is likely the retinal ganglion, bipolar, and photoreceptor cells.91,95 The exact mechanism of quinine toxicity is unidentified, but some have suggested that it may act as an acetylcholine antagonist and disrupt cholinergic transmission in the retina.96
Clofazimine
Clofazimine is a red phenazine dye that has been used to treat dapsone-resistant leprosy, psoriasis, pyoderma gangrenosum, discoid lupus, and more recently, Mycobacterium avium-complex infections in AIDS patients. With treatment over several months, clofazimine crystals may accumulate in the cornea. Two cases of bull’s-eye maculopathy with pigmentary retinopathy (Fig. 89.13) have been reported in AIDS patients with doses of 200 to 300 mg/day (total dose, 40–48 g).97,98 Visual acuity was mildly affected, with reduced scotopic, photopic, and flicker ERG amplitudes. Cessation of treatment may result in the clearance of the corneal deposits but does not appear to affect the retinopathy.
Dideoxyinosine (DDI)
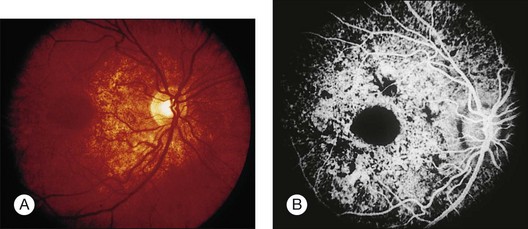
A midperipheral pigmentary retinopathy has been noted in three children with AIDS receiving high-dose therapy with the antiviral 2’, 3’-dideoxyinosine.99 The cases were associated with ERG and EOG changes. The retinal toxicity stabilized after discontinuation of the medication. Several cases have now been described involving adult patients as well. A very similar midperipheral pattern of RPE abnormality has been noted (Fig. 89.14).
Deferoxamine
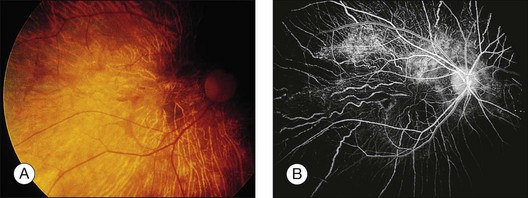
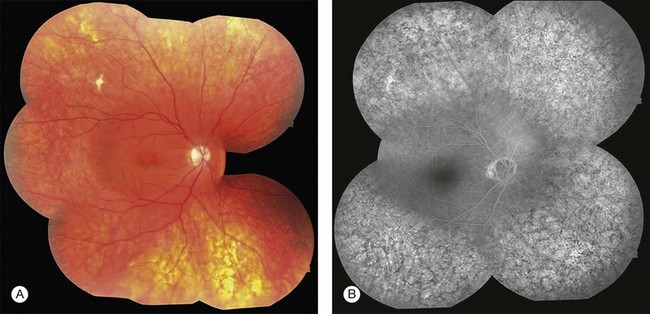
Intravenous (IV) and subcutaneous (SQ) administration of deferoxamine has been used to treat patients who require repeated blood transfusions and subsequently develop complications of iron overload. High-dose IV and SQ therapy has produced visual loss, nyctalopia, peripheral and central field loss, and reduced ERG amplitudes and EOG ratios.100,101 The fundus examination can be normal initially, or there may be a faint graying of the macula.102 Pigmentary changes in the macula and periphery develop within a few weeks and are particularly highlighted by fluorescein angiography103 (Fig. 89.15). Return of visual function occurs with cessation of therapy. Deferoxamine chelates many metals other than iron, and it is possible that the mechanism of toxicity may involve the removal of copper from the RPE.100 Histopathologic changes occur primarily in the RPE and include loss of microvilli from the apical surface, patchy depigmentation, vacuolation of the cytoplasm, swelling and calcification of mitochondria, and disorganization of the plasma membrane.104
Corticosteroid preparations
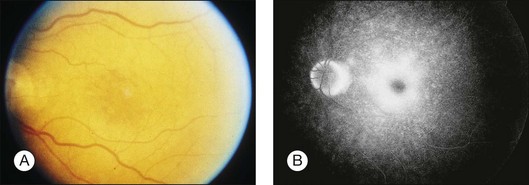
The vehicles of several common corticosteroid preparations have been shown to cause retinal necrosis when inadvertently injected into the eye105,106 (Fig. 89.16). The corticosteroids themselves probably have a minimal toxic effect on the retina.107 Celestone Soluspan, with its vehicle benzalkonium chloride, and Depo-Medrol, with myristyl gamma-picolinium chloride, caused the most extensive retinal damage in an experimental study comparing several depot steroids.108 If one of these agents is inadvertently injected, immediate surgical removal should be instituted.
Cisplatin and BCNU (carmustine)
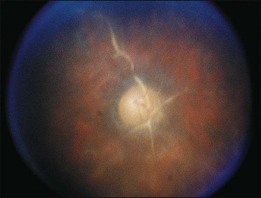
Cisplatin and BCNU are used for the treatment of malignant gliomas and metastatic breast cancer. Three different types of retinal toxicity have been reported with these agents. One type of change consists of a pigmentary retinopathy of the macula with markedly decreased visual acuity and frequently abnormal electrophysiologic testing. This pigmentary change has been reported after administration of combined intra-arterial cisplatin and BCNU and with cisplatin alone for malignant glioma.109,110 These findings probably are the result of platinum toxicity of the retina. Severe bilateral visual loss was reported after intravenous cisplatin in a patient that received four times the intended dose for treatment of lymphoma.111 Later histology showed a splitting of the outer plexiform layer.
A second type of retinopathy has been described and consists of cotton-wool spots, intraretinal hemorrhages, macular exudate, and optic neuropathy with disc swelling. This was reported in the setting of high-dose chemotherapy with cisplatin, cyclophosphamide, carmustine, and autologous bone-marrow transplantation for metastatic breast cancer.112 The third type of change involves a vascular retinopathy or optic neuropathy, which can include arterial occlusion, vasculitis, and papillitis. This has been seen in approximately 65% of patients receiving intra-arterial BCNU alone or combined with cisplatin for malignant glioma.110 These fundus changes are associated with a profound visual loss that begins about 6 weeks after the start of therapy. Other ocular effects may include orbital pain, chemosis, secondary glaucoma, internal ophthalmoplegia, and cavernous sinus syndrome. Injection of medication above the ophthalmic artery can still result in toxicity.113 The visual loss usually is progressive, and no treatment is known.
Miscellaneous agents
Overdose of potassium iodate, an iodized salt used for iodine supplementation in areas endemic for goiter, has been shown to cause profound visual loss and extensive fundus pigmentary abnormalities.114 Fluorescein angiography reveals RPE window defects and ERG and VEP testing shows marked impairment of retinal function. Visual acuity may improve slowly over several months. There have been two cases of pigmentary retinopathy with diminished ERG amplitudes reported after administration of denileuken diftitox.115
Vascular damage
Talc
A characteristic retinopathy consisting of small, white, glistening crystals concentrated in the end arterioles of the posterior pole has been described in intravenous (IV) drug abusers116–118 (Fig. 89.17). These addicts crush oral medications such as methylphenidate hydrochloride (Ritalin) or methadone HCl and then create an aqueous suspension by adding water and heating the mixture. The solution is subsequently drawn up into a syringe, with occasional attempts at filtering the mixture with cotton fibers, gauze, or cigarette filters. These oral medications contain talc (hydrous magnesium silicate) as inert filler material; after IV administration, talc particles embolize to the pulmonary vasculature, where the larger particles are trapped. After repeated injections over months to years, collateral vasculature develops, allowing the particles to enter the systemic circulation and embolize to other organs, including the eye. Even before shunt development, particles smaller than 7 µm can traverse the pulmonary capillary bed and enter the retinal circulation.119
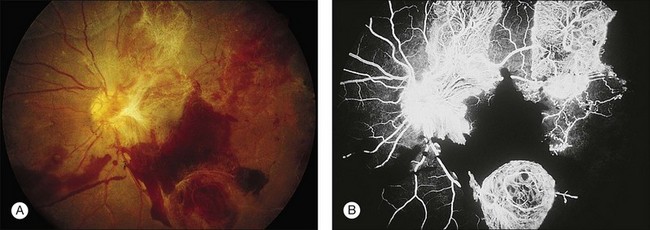
Once a large number of talc particles lodge in the small arterioles of the retinal vasculature, a characteristic picture of an ischemic retinopathy begins to develop. Capillary nonperfusion, microaneurysm formation, cotton-wool spots, and venous loops can all be seen.120 In severe cases optic disc and peripheral neovascularization and vitreous hemorrhage can develop121,122 (Fig. 89.18). An experimental model of talc retinopathy in monkeys has demonstrated with light and electron microscopic techniques that the vascular abnormalities induced are very similar to other ischemic retinopathies seen in humans, such as sickle cell and hypertensive retinopathy.123–125
Oral contraceptives
Oral contraceptives have been implicated in some cases of central retinal vein occlusion (CRV), retinal and cilioretinal artery obstruction, and retinal edema occurring in young women.126–132 The synthetic estrogen and progesterone contained in contraceptive pills are thought to adversely effect coagulation factors and induce a hypercoagulable state leading to thromboembolic complications. Most of the studies reporting ocular complications are from the 1960s and 1970s, when the estrogen concentrations used in “the pill” were much higher (Fig. 89.19). Some recent prospective studies have failed to show an increased incidence of ocular complications with the drug, though one large study showed an increase in “retinal vascular findings”.133–135
Aminoglycoside antibiotics
Retinal toxicity from aminoglycoside antibiotics has been reported after inadvertent intraocular injection of massive doses, intravitreal injection for bacterial endophthalmitis, prophylactic intravitreal injection after pars plana vitrectomy, prophylactic subconjunctival injections after routine ocular surgery, and with the use of small amounts in the infusion fluid during cataract extraction.136–139 Gentamicin is the most toxic antibiotic in the aminoglycoside family, followed by tobramycin and amikacin.140 Massive doses result in early superficial and intraretinal hemorrhages, retinal edema, cotton-wool patches, arteriolar narrowing, and venous beading139 (Fig. 89.20). Fluorescein angiography reveals severe vascular nonperfusion in the acute stages. Visual loss is profound, and late rubeosis iridis, neovascular glaucoma, pigmentary retinopathy, and optic atrophy are common. Intravitreal injection of smaller doses thought to be safe for the eye (100–400 µg) can still cause toxicity with less severe fundus changes.137–139 The major preservatives found in injectable gentamicin (methylparaben, propylparaben, sodium bisulfite, and edetate disodium) likely play an additive role in its ocular toxicity.
A number of factors appear to affect the extent of toxicity observed with similar doses of these medications. Peyman found that retinal toxicity could be enhanced with an intravitreal injection directed at the posterior pole with the bevel of the needle pointed toward the retina, and Zachary and Forster demonstrated that an increased rate of injection during intraocular administration could also increase the retinal toxicity observed.141,142 One investigator stated that eyes that have undergone a previous pars plana vitrectomy are at greater risk for gentamicin toxicity, but an experimental model has shown no difference between eyes that had cataract extraction alone compared with those that underwent lensectomy and vitrectomy.143,144 Finally, increased ocular pigmentation protects the rabbit retina from aminoglycoside toxicity and may explain some of the wide variability seen with intraocular exposure in humans.145,146
Although clinical aminoglycoside toxicity appears to affect the retinal vasculature primarily, pathologic studies have revealed that gentamicin in small doses causes the formation of abnormal lamellar lysosomal inclusions in the RPE, and larger doses cause increasing amounts of retinal necrosis, first of the outer then inner segments.147–150 Histologically, vessel closure appears to result from granulocytic plugging.
Prevention of aminoglycoside toxicity can be accomplished by abandoning the use of these medications as routine prophylaxis following intraocular surgery, eliminating them from intraocular infusion fluids used in vitrectomy and cataract surgery, and using alternative medications for the treatment of bacterial endophthalmitis. Animal studies have demonstrated that thinned sclera alone without perforation can result in markedly elevated intraocular gentamicin levels after subconjunctival injection.151 If inadvertent intraocular injection does occur, immediate pars plana vitrectomy with posterior segment lavage should be performed.152,153 Since there is some evidence that gravity plays a role in the predilection of gentamicin-induced toxicity for the macula, the patient should be placed upright as soon as possible after surgery.154
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


