Chapter 17 Disorders of the eye as a whole
Anophthalmos and microphthalmos
Anophthalmos and microphthalmos are rare, with an incidence of between 2 and 19 per 100 000 live births, respectively.1–3 They are often associated with systemic malformations. Although reported risk factors include maternal age over 40 years, multiple births, low birth weight, and low gestational age, there is no unifying causation, and clustering of cases, which might suggest an environmental cause, probably does not occur.1,2,4,5
Anophthalmos
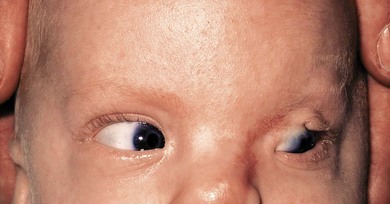
Anophthalmos is when the eye is non-existent (Fig. 17.1) or, more commonly, when it is not visible and a tiny cystic remnant of the eye is identified on pathology. The term, “clinical anophthalmos,” emphasizes that there is a spectrum in which anophthalmos merges with microphthalmos. True anophthalmos is sometimes associated with absence of the optic nerve and chiasm, as in some patients with SOX2 mutations.6
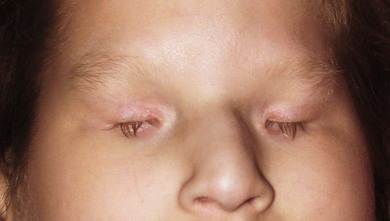
Fig. 17.1 Bilateral anophthalmos in a girl with a SOX2 mutation. There was absence of all visual pathways.
Secondary abnormalities of the orbit occur, with orbital growth universally retarded to some extent. Extraocular muscles may be absent and the optic foramen size is often decreased. The conjunctival sac may be small. Although the absence of a developing eye does not affect the initial development of a bony orbit,7 the growth of the orbit is highly influenced by the presence or absence of an eye. The mean axial length of the human full-term neonatal and adult eyes are approximately 17 and 23.8 mm, respectively,8 with the normal neonatal eye measuring 70% of the adult size.9 Orbital volume increases dramatically during the first 3 years of life, especially in the first year.
While orbital volume cannot be assessed using plain radiographs, the horizontal and vertical sizes of the orbital openings from the orbital rim can be measured and are reduced in adults with congenital anophthalmos, or in those enucleated within the first year of life. This retardation of orbital growth is halved when an orbital implant is used, and the severity of the overall reduction in volume diminishes if the insult occurs at a later date. Orbital growth appears to be complete by the age of 15 years; subsequent enucleation will not result in appreciable size difference.10
Many causes for anophthalmos have been proposed; these merge with the causes of microphthalmos (see next section). Bilaterality and increased severity imply an early teratogenic or genetic developmental event.11 In isolated cases, inheritance may be autosomal dominant12 or autosomal recessive.13 In families with clear Mendelian inheritance, ocular pathology may be asymmetric or even unilateral. Mutations in SOX2 are one of the more common causes of anophthalmos.14
Microphthalmos
The volume of a microphthalmic eye is reduced. Often, clinical suspicion arises from a corneal diameter less than 10 mm in adults. Although microphthalmos is usually associated with a small cornea, there may be microphthalmos with a normal cornea, and microcornea without microphthalmos.15,16 Ultrasonographic determination of an axial length less than 21 mm in an adult or 19 mm in a 1-year-old child substantiates a diagnosis of microphthalmos.17 This represents a reduction of 2 standard deviations or more below normal.
Bilateral microphthalmos18 occurs in approximately 10% of blind children.19 The effect of microphthalmos on vision depends on whether it is bilateral, the severity of the microphthalmos, and associated ocular malformations – specifically, the degree of retinal maldevelopment, horizontal corneal diameter, and the presence or absence of cataract and coloboma.20
Microphthalmos may be simple (without other ocular defects) or complex (associated with anterior segment malformations, cataracts, retinal or vitreous disease, or more complex malformations).17,21 It can be further divided into colobomatous (Fig. 17.2) and non-colobomatous on the basis of associated uveal abnormalities.15,22 The association between eye growth and closure of the fetal fissure is important since closure of the cleft is completed early in development.23
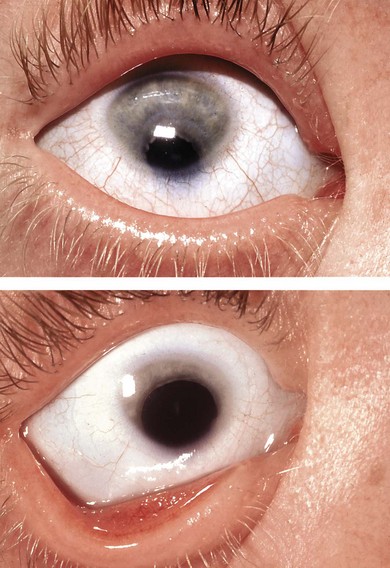
Microphthalmos represents a non-specific growth failure of the eye in response to prenatal insults and genetic defects. Inadequate postnatal growth, secondary to decreased size of the optic cup, altered vitreous composition, and low intraocular pressure, may play a role in the pathogenesis of simple micophthalmos.21 Posterior segment abnormalities and complex microphthalmos may be secondary to inadequate production of secondary vitreous.17 Microphthalmos can be classified according to mode of inheritance, environmental causes, chromosomal aberration, as well as syndromic associations that have additional systemic abnormalities. (For a complete search of inherited conditions associated with microphthalmos and anophthalmos, see http://www.ncbi.nlm.nih.gov/omim.)7,15
Inherited isolated microphthalmos
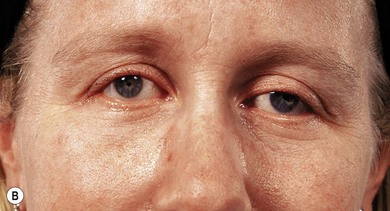
Although most cases are sporadic,24,25 some are autosomal dominant.26 Some families (Fig. 17.3) have dominant inheritance of colobomatous microphthalmos, with variable expression with extreme microphthalmos at one end of the spectrum and coloboma at the other. A high rate of consanguinity suggests an autosomal recessive inheritance in some cases.13,27 X-linked recessive inheritance occurs, sometimes with mental retardation.28
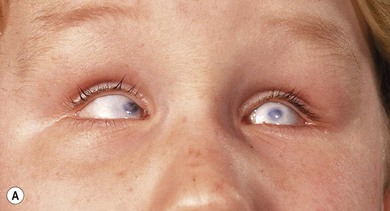
Microphthalmos with orbital cyst
This form of microphthalmos presents with a bulge behind the lower lid from birth (Fig. 17.4A). It is secondary to failure of optic fissure closure and the protrusion of a cyst from the coloboma. It has been confused with a congenital cystic eye,29 but the two are different. In the latter, the eye is replaced by a cystic structure and there is no lens or other normal eye structures. In microphthalmos with cyst, the small eye often cannot be seen and a neoplasm may be suspected. The cyst usually communicates with the eye.30–32 Presentation may be as an orbital mass distending the lids and hiding the eye, or as proptosis in which a microphthalmic eye is visible. Ultrasonography and CT or MRI scanning aid in diagnosis (Fig. 17.4B).31 Most cases of microphthalmos with cyst are sporadic; familial cases have been reported, with presumed autosomal recessive inheritance.30,33,34
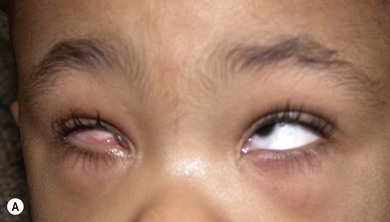
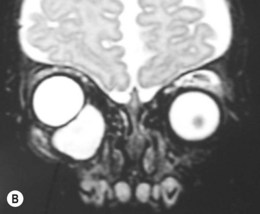
Management is initially conservative, especially for small cysts. Large cysts may be managed with repeated aspiration or by surgical removal.31,35–37 If the cyst is not growing rapidly, it may be left in place until some orbital growth is achieved. Because of the communication of the cyst with the eye, removal of the cyst may deflate the microphthalmic eye necessitating its removal.
Microphthalmos with cryptophthalmos
Cryptophthalmos implies a varying degree of skin covering the eyeball, with variable cutaneous adhesions to the cornea.38 It is usually bilateral with a variable degree of severity. Unilateral cases have been described.
Francois described three subgroups of cryptophthalmos:39
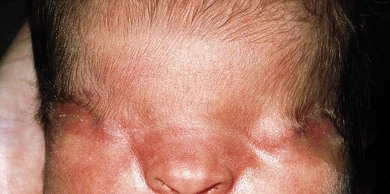
1. Complete cryptophthalmos (Fig. 17.5): the lids are replaced by a layer of skin without lashes or glands that is fused with the microphthalmic eye without a conjunctival sac. Normal electrophysiological responses have been recorded.40
2. Incomplete cryptophthalmos (Fig. 17.6): the lids are colobomatous (often medially) or rudimentary and there is a small conjunctival sac. The exposed cornea is often opaque.
3. An abortive form: the upper lid is partly fused with the upper cornea and conjunctiva and may be colobomatous.38 The globe is often small.
A fourth autosomal dominant type exists in which the upper lid is very tall and fused with the lower one at the margins. There is a normal complement of lashes. A dimple in the upper lid indicates where it is attached to the underlying eyeball.41
The systemic associations with cryptophthalmos and microphthalmos include nose deformities, cleft lip and palate, syndactyly, abnormal genitalia, renal agenesis, mental retardation, and many others.38,42,43
Microphthalmos with ocular and systemic malformations
Other eye malformations and systemic abnormalities are frequent in microphthalmos. Numerous syndromes associated with microphthalmos have been reported (http://www.ncbi.nlm.nih.gov/omim).44
Microphthalmos with ocular abnormalities
Microphthalmos is a non-specific response to a wide variety of influences. It occurs with many severe eye diseases, including anterior segment malformations such as Peters’ anomaly or cataracts, especially in the context of a chromosomal abnormality,45 persistent hyperplastic primary vitreous (PHPV),46 and multisystem syndromes such as the oculodentodigital syndrome.47 Microphthalmos may be secondary to severe, widespread intraocular disease including retinopathy of prematurity, retinal dysplasia, retinal folds,48 retinal degeneration and glaucoma. A three-generation family has been described with aniridia, anophthalmos, and microcephaly.49 Coloboma is the most common associated ocular malformation with microphthalmos and is found in many microphthalmos syndromes.50,51
Microphthalmos with systemic malformations
Up to 50% of patients with anophthalmos and microphthalmos have associated systemic abnormalities.9,52 Many patients with microphthalmos-associated syndromes, especially chromosomal disorders, are mentally retarded28,53,54 or have cleft palate with and without macrosomia.55
The most common syndromic cause of colobomatous microphthalmos is the CHARGE syndrome (Coloboma, Heart abnormalities, Atresia of the choanae, Retardation of growth and development, Genitourinary abnormalities, and Ear/hearing abnormalities).50 Patients can have cranial nerve abnormalities such as facial nerve palsy, craniofacial clefting, dysphagia/esophageal abnormalities, duplication of the thumb, and congenital brain abnormalities, particularly of the forebrain.56,57 Although most cases are sporadic, autosomal dominant transmission has been reported.58 Mutations in the CHD7 gene are responsible for 60% of CHARGE cases, with possible genotype–phenotype correlation.59,60 CHD7 encodes a putative chromodomain protein widely expressed in the neuroectoderm and in neural crest cells during human development.61 Mutation in the SEMA3E gene can result in CHARGE syndrome.62
The Temple-al-Gazali syndrome (Fig. 17.7) also referred to as X-linked dominant microphthalmos with linear skin defects (MLS) syndrome or the microphthalmos, dermal aplasia, and sclerocornea (MIDAS) syndrome results from a deletion of Xp22.2-pter.63,64 Patients have linear, irregular areas of skin aplasia especially of the head and neck, microphthalmos with variable sclerocornea, and, sometimes, abnormal intelligence.65–67 The condition is lethal in XY males.
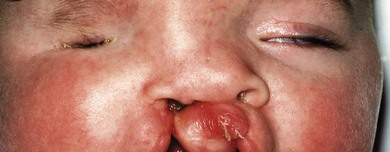
Fryns “anophthalmos plus” syndrome is microphthalmos or anophthalmos, cleft lip or palate, and sacral neural tube defect.68 The branchio-oculofacial syndrome combines a broad nose with large lateral pillars, branchial sinuses, and orbital cysts.69,70 Other microphthalmos syndromes with facial defects include fronto-facio-nasal dysplasia (Fig. 17.8),71 and the cerebro-oculo-nasal syndrome in which there is an asociation of anophthalmos/microphthalmos, abnormal nares, and central nervous system anomalies.72
In Delleman’s syndrome there is an association of skin tags, punched-out lesions of the skin on the ears and elsewhere, mental retardation, hydrocephalus, brain malformations, and orbital cysts.73,74
Microphthalmos has been described in patients with growth retardation, microcephaly, brachycephaly, oligophrenia syndrome (GOMBO syndrome).75
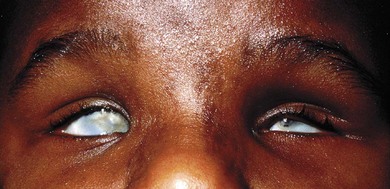
The eyes can be quite small in some patients with the oculo-dento-digital syndrome, characterized by bilateral digital anomalies (Fig. 17.9) with cutaneous syndactyly of fingers and camptodactyly,47,76 thin nose with hypoplastic alae nasi and small nares, partial dental agenesis, enamel hypoplasia, and glaucoma.77,78
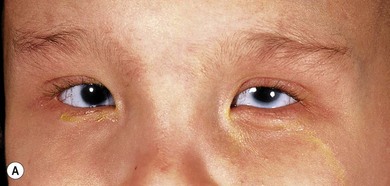
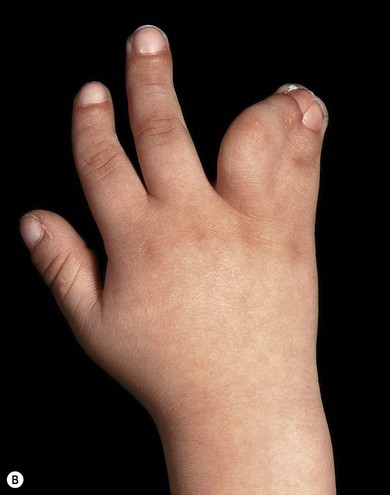
Patients with the less common recessive variety are more likely to have microphthalmos.79 The syndrome results from mutations in Connexin 43. Iris cysts and anomalous retinal development may occur.80
Waardenburg’s recessive anophthalmos syndrome includes microphthalmos with syndactyly, oligodactyly, and other limb defects and mental retardation.81
Patients with the X-linked recessive Lenz’ microphthalmos syndrome have microphthalmos with mental retardation, malformed ears, and skeletal anomalies.51,82,83 The gene maps to Xq27-q28 but has not been identified yet.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


