Diseases of the Retina and Vitreous
Eric D. Weichel
James F. Vander
William Tasman
William E. Benson
X-LINKED RECESSIVE RETINOSCHISIS
X-linked recessive retinoschisis is an inherited ocular disorder that occurs in males (1). It is characterized by degeneration of the vitreous and splitting of the retina at the level of the nerve fiber layer. The most common finding involves the macula and consists of a petaloid configuration. Usually there are numerous folds that radiate in a spoke-wheel configuration (Fig. 14.1). This may give the clinical appearance of cystoid macular edema, but the macula does not stain with fluorescein. When seen in a young boy, this sign should alert the observer to the possibility of the peripheral changes seen in X-linked retinoschisis. Peripheral retinoschisis, seen 50% of the time, is more common in the inferotemporal quadrant. The schisis is always bilateral but may be asymmetric. The anterior limit of the retinoschisis seldom extends to the ora serrata, and the posterior limit may extend to the optic disc. Nerve fiber layer breaks are common and appear as large round or oval holes (Fig. 14.2). In some eyes the nerve fiber layer breaks are so large that only remnants of the nerve fiber layer remain. Often, bridging retinal blood vessels are present, and there is hemorrhage into the vitreous. When vitreous hemorrhaging occurs, some patients develop dragging of the retina. Most recently, ocular coherence tomography (OCT) has demonstrated a characteristic finding of a wide hyporeflective space between the thin reflective outer layer and the thicker, more reflective inner retinal layers (Fig. 14.3) (2).
Vitreous veils and strands may also be present. The electroretinogram (ERG) often shows a subnormal b-wave in conjunction with a normal a-wave. Color vision abnormalities parallel the degree of foveal involvement. The results of electrooculogram (EOG) and dark adaptation tests are usually normal. Most commonly, patients are first seen because of decreased vision. The visual acuity on presentation is usually between 20/70 and 20/100 mmHg. This often (but not always) deteriorates up until age 20 years, reaching the 20/200 range. Other presenting symptoms are vitreous hemorrhage, retinal detachment, and strabismus.
The natural history of retinoschisis is that of a stationary or slowly progressive disease. The most important complications are vitreous hemorrhage and retinal detachment. It is important to realize that progression of X-linked retinoschisis may be followed by spontaneous partial regression, and that fluctuations in the appearance of the fundus are common during the first few years of life.
Differential diagnoses include retinal detachment, persistent fetal vasculature (PFV), Goldmann-Favre disease, retinitis pigmentosa (RP), Norrie disease, Stickler’s syndrome, and (because of occasional dragged retinas) retinopathy of prematurity (ROP) and familial exudative vitreoretinopathy (FEVR).
Retinal detachment in a child may be differentiated from X-linked retinoschisis because the latter is always bilateral. In addition, retinal detachment, unlike X-linked retinoschisis, usually extends to the ora serrata.
In some cases of PFV, extensive hyaloid remnants that are adherent to the disc and inferior retina may contract and cause an inferior retinal detachment, with or without visible retinal breaks. This condition is generally unilateral and associated with microphthalmos, and is neither familial nor hereditary.
Goldmann-Favre vitreoretinal degeneration is transmitted as an autosomal recessive trait. Although peripheral retinoschisis is often present, the disease is also characterized by night blindness and fundus changes resembling those of RP.
Stickler’s syndrome is transmitted as an autosomal dominant trait. Elevation of the retina is attributable to rhegmatogenous retinal detachment rather than retinoschisis. Additional ophthalmologic and systemic features help to distinguish this entity from X-linked retinoschisis.
Although dragging of the retina may occur in ROP and FEVR, the additional fundus features of these two entities are distinct and rarely confused with X-linked retinoschisis. In addition, ROP and FEVR can usually be identified because of a history of prematurity in the case of ROP and autosomal dominant inheritance with respect to FEVR.
As long as X-linked retinoschisis is not accompanied by rhegmatogenous retinal detachment, no treatment is indicated. Recurrent vitreous hemorrhages are usually best treated conservatively, but vitrectomy occasionally becomes necessary because of the presence of organized vitreous membranes leading to retinal detachment.
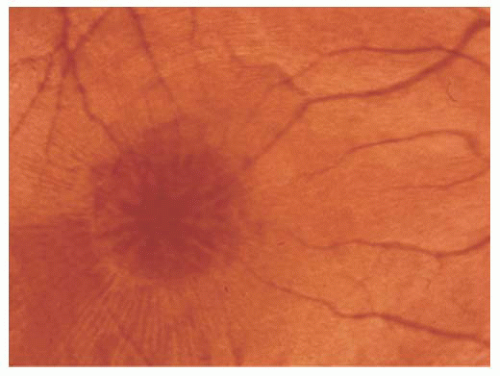 FIGURE 14.1. Foveoschisis with typical retinal cysts in a petaloid configuration and radial striae in X-linked retinoschisis. |
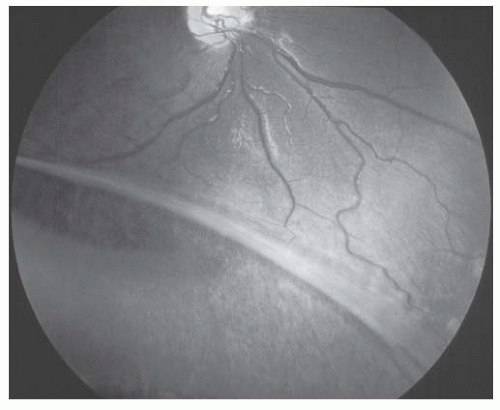 FIGURE 14.2. Peripheral nerve fiber layer dehiscence in X-linked retinoschisis. |
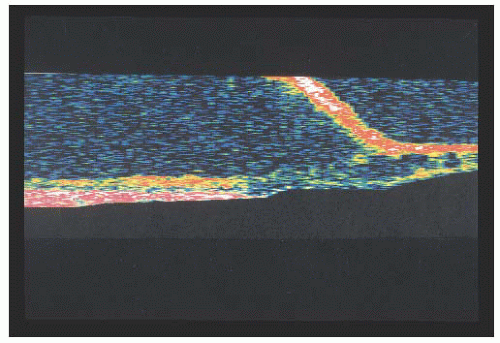 FIGURE 14.3. Ocular coherence tomography (OCT) of X-linked retinoschisis showing schisis of nerve fiber layer. |
X-linked retinoschisis has a prevalence ranging from 1:5,000 to 1:25,000. Female carriers of X-linked retinoschisis generally do not show any ocular abnormalities, although peripheral retinal alterations similar to those found in affected males have been reported (3). The X-linked retinoschisis gene (XLRS1) is located on the distal short arm of the × chromosome (Xp22) (4). DNA analysis can reveal evidence of the carrier state and is of use when performing genetic counseling.
HEREDITARY VITREORETINOPATHY WITH SYSTEMIC FINDINGS
Stickler’s Syndrome
Stickler and associates (5) described an autosomal dominant, progressive arthro-ophthalmopathy associated with high myopia, optically empty vitreous, and retinal detachment. Stickler’s syndrome is the most common disorder associated with high myopia and retinal detachment. Systemic findings include midfacial flattening (Fig. 14.4), cleft palate, micrognathia, glossoptosis, hearing loss, and skeletal dysplasia. The ocular findings include an optically empty vitreous with bands. Myopia is common. Lattice degeneration is present and often radial and perivascular (Fig. 14.5). There is a high
incidence of retinal breaks, which may be multiple or giant retinal tears. Cataracts and glaucoma are often present.
incidence of retinal breaks, which may be multiple or giant retinal tears. Cataracts and glaucoma are often present.
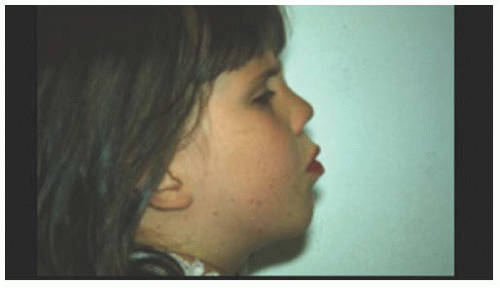 FIGURE 14.4. Flattened facies in a young patient with Stickler’s syndrome. |
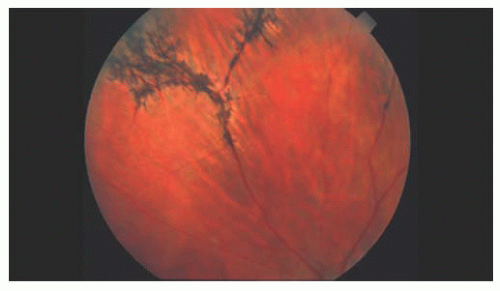 FIGURE 14.5. Radially oriented lattice degeneration in a patient with Stickler’s syndrome. |
Treatment of retinal detachment in Stickler’s syndrome is difficult because of posterior retinal breaks and a high incidence of proliferative vitreoretinopathy. Prophylactic laser treatment to areas of lattice degeneration and retinal breaks may reduce the risk of detachment.
Stickler’s syndrome has been linked to mutations in the type II procollagen (COL2A1) gene. A polymerase chain reaction assay is available to assist in genetic counseling (6).
HEREDITARY VITREORETINOPATHY WITHOUT SYSTEMIC FINDINGS
Wagner’s Syndrome
Wagner’s syndrome (Wagner’s hereditary vitreoretinal degeneration) (7) is similar to Stickler’s syndrome but without any systemic abnormalities. These patients have myopia, an optically empty vitreous cavity, preretinal avascular membranes, perivascular pigmentation, retinal degeneration, and progressive chorioretinal atrophy. They also develop lenticular changes between the ages of 20 and 40 years. Wagner’s syndrome is autosomal dominant and has been localized to chromosome 5q13-q14 (8). Patients with Wagner’s syndrome infrequently develop retinal detachment compared with a much higher incidence of retinal detachments in patients with Stickler’s syndrome.
Goldmann-Favre Disease
Goldmann-Favre disease (9) is inherited in an autosomal recessive manner. It is characterized by night blindness with absent or diminished ERG response, foveal and peripheral retinoschisis, pigment changes resembling RP, and progressive decreased visual function (Fig. 14.6) (6). As in Stickler’s syndrome, the vitreous is liquefied with vitreous strands and veils. Retinal detachments and cataract formation are common in this condition. Retinal detachments have a guarded prognosis for successful repair; therefore, asymptomatic breaks should be treated prophylactically before detachment. The enhanced S cone syndrome is a variant of Goldmann-Favre disease with night blindness and foveal cystic changes without the vitreous abnormalities.
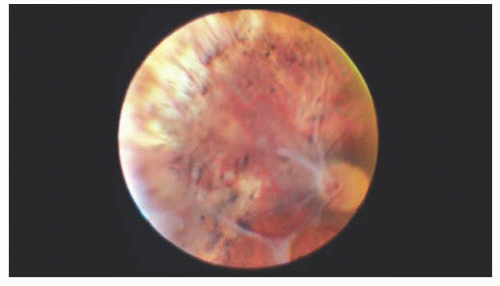 FIGURE 14.6. Preretinal membranes and retinal pigmentary changes in a 34-year-old woman with Goldmann-Favre disease. |
STARGARDT’S DISEASE (FUNDUS FLAVIMACULATUS)
Stargardt’s disease is most often an autosomal recessive condition that usually appears between 8 and 14 years of age. It is bilateral, slowly progressive, and sometimes associated with macular degeneration (10). Characteristically, the foveal reflex is absent or grayish in color. Pigmentary spots sometimes develop in the macular area and may accumulate irregularly. Yellowish-white pisciform flecks may be visible in the deep retina or retinal pigment epithelium (RPE) (Fig. 14.7). They are typically seen in the posterior pole but can extend out to the equator. Eventually, in some cases, a circular area of depigmentation and chorioretinal atrophy of the macula follow (Figs. 14.8 and 14.9). In the early stages of the disease, the loss of central vision may be out of proportion to the appearance of the fundus. Fluorescein angiography may reveal abnormalities, particularly a dark fundus (the so-called silent choroid) (11) before any fundus abnormalities become apparent. Fluorescein angiography of the flecks may reveal hypofluorescence, presumably because of blockage. Later, some areas may hyperfluoresce because of damage to the RPE. Sometimes, the entire choroid may show blockage on fluorescein angiography.
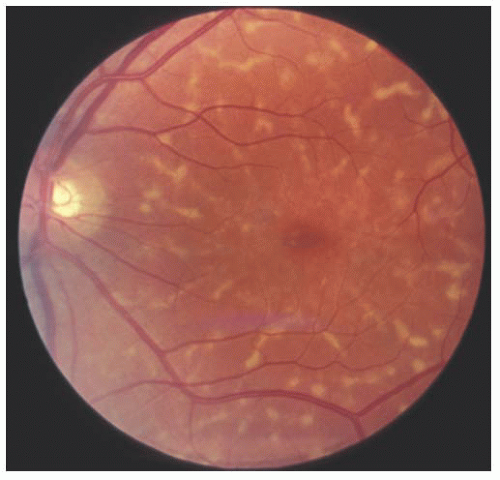 FIGURE 14.7. Typical pisciform lesion of fundus flavimaculatus. The dark fundus and pisciform lesions are the result of excessive amounts of lipofuscin in the pigment epithelium. |
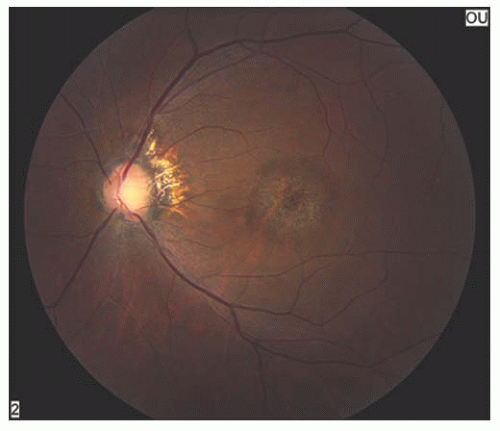 FIGURE 14.8. Typical retinal pigment epithelium (RPE) atrophy in a bull’s-eye pattern in a patient with Stargardt’s disease. |
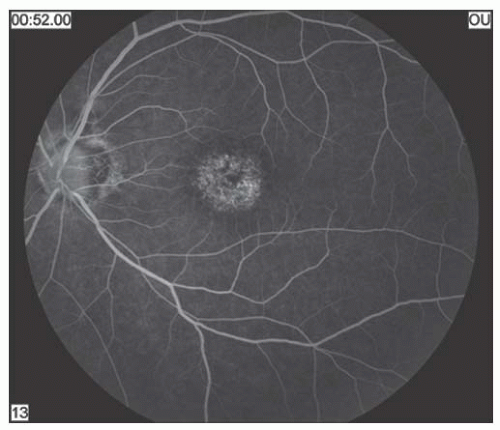 FIGURE 14.9. Corresponding fluorescein angiography with centra hyperfluorescence. |
The evolution is slow, symmetric, and progressive, and the disease is usually well established by age 30 years, with vision in the 20/200 range. Late in life, large areas of chorioretinal atrophy may develop (12).
The disease was first described by Stargardt in 1909. Fundus flavimaculatus was described independently as a separate entity. Today, however, Stargardt’s disease and fundus flavimaculatus are thought to have a common cause and to represent different parts on the spectrum of a single disease. Stargardt’s disease is caused by a mutation of the ABCR gene located on the short arm of chromosome 1 (13). Other sites have been identified in patients with the autosomal dominant form. The mechanism of photoreceptor death has been postulated (14) with histopathology revealing the accumulation of lipofuscin within the RPE. Stargardt’s disease refers to predominantly macular involvement, and fundus flavimaculatus refers to more peripheral involvement. In fundus flavimaculatus, the vision is near normal unless macular involvement develops.
BEST’S VITELLIFORM DEGENERATION
In 1905, Best reported eight members of one family with an interesting macular dystrophy, now called Best’s vitelliform degeneration (15). The transmission in this disease is autosomal dominant, but there may be variable expressivity. Vitelliform macular degeneration has a distinctive appearance characterized by a sharply defined discoid formation in, or immediately adjacent to, the macula (Fig. 14.10). The disc is usually yellow-orange or pinkish yellow and varies in size from 0.5 to 4 disc diameters. The abnormality is subretinal and resembles the yolk of a poached egg (16). It is usually diagnosed between 5 and 15 years of age and is bilateral, although unilateral cases have been reported. Multiple vitelliform lesions in the same eye have also been described. The condition is very slowly progressive. The vision is usually normal or mildly reduced at this stage. Gradually the homogeneous contents of the vitelliform disc may “scramble,” giving an irregular yellow lesion, eventually leaving abnormal pigmentation and chorioretinal atrophy (Fig. 14.11). The appearance at that point is often indistinguishable from that associated with other types of macular degeneration. Vision loss develops from these atrophic changes or, in some cases, a choroidal neovascular membrane. These macular changes can also be assessed with the OCT (17).
Best’s dystrophy of the macula is present at birth and if no change is visible postnatally, the clinical signs will not develop later.
ERG results are normal, as are the peripheral visual fields. Central scotomata cannot be elicited in eyes with normal visual acuity but are present late in the disease. Dark adaptation is normal. The EOG, however, is always
abnormal in patients with vitelliform macular degeneration, even in those who do not express the disease clinically. Thus, EOG testing is helpful diagnostically and in genetic counseling, because unaffected carriers have a 50% chance of passing the condition to their offspring. Carriers who have a normal ophthalmologic examination will have a subnormal EOG (18).
abnormal in patients with vitelliform macular degeneration, even in those who do not express the disease clinically. Thus, EOG testing is helpful diagnostically and in genetic counseling, because unaffected carriers have a 50% chance of passing the condition to their offspring. Carriers who have a normal ophthalmologic examination will have a subnormal EOG (18).
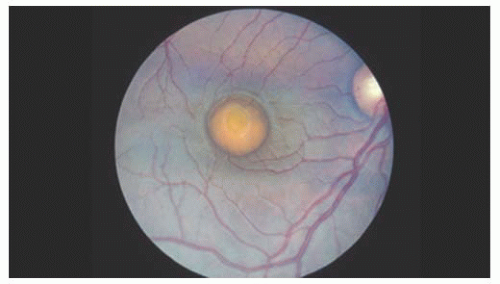 FIGURE 14.10. Fried-egg appearance of a typical vitelliform macular degeneration. |
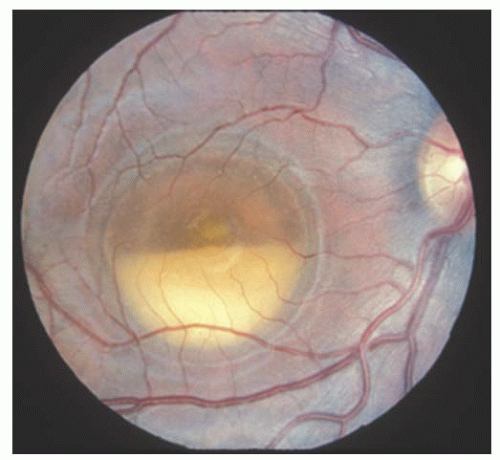 FIGURE 14.11. Pseudohypopyon stage of Best’s disease resulting from a fluid level within a cystic space. |
The pathogenesis of vitelliform degeneration is uncertain. The gene causing Best’s disease has been localized to 11q13 with an identified encoded protein called bestrophin, which has an unknown function (19).
STATIONARY FORMS OF CONGENITAL NIGHT BLINDNESS
The stationary forms of congenital night blindness are congenital stationary night blindness, Oguchi’s disease, and fundus albipunctatus. These diseases should be considered in the differential diagnosis of early-onset night blindness that is not progressive. All of the former differ from the progressive disorders, such as RP, Goldmann-Favre disease, and gyrate atrophy.
Congenital Stationary Night Blindness
Congenital stationary night blindness exhibits three modes of inheritance: (a) X-linked (most common), (b) autosomal dominant, and (c) autosomal recessive. Molecular genetic testing has found numerous mutations in genes encoding proteins of photoreceptors or the RPE (20). Color vision and visual fields characteristically are normal. Visual acuity is normal or mildly reduced. The fundi are entirely normal. Histopathologically the retina is normal. Dark adaptation reveals a reduced retinal sensitivity, and ERG shows a decreased scotopic response with a normal photopic response. The defect is caused from a failure of communication between the proximal end of the photoreceptor and the bipolar cell. No Purkinje shift in relative luminosity curves is seen. Initially, the disease can be confused with early-onset RP, but the lack of progression with the former serves to distinguish these two entities.
Oguchi’s Disease
Oguchi’s disease, another stationary form of congenital night blindness, is usually diagnosed by a combination of two readily observable phenomena. First is the unusual color of the fundus, which has been described as various shades of gray-white to yellow. The abnormal color may be limited to a small section of the mid-periphery or extend throughout the entire fundus in a discontinuous or homogeneous pattern. The second unique characteristic of Oguchi’s disease is the Mizuo phenomenon (21), which is a change in the color of the fundus in the dark-adapted state (Fig. 14.12). When light is prevented from entering the eye, the color of the fundus changes from the light shade, seen initially, to a reddish, more normal appearance. The time needed to elicit this change varies among patients. Dark adaptation testing reveals a prolonged dark adaptation time and normal retinal sensitivities. ERG testing reveals a decreased scotopic response, which may revert to normal during prolonged dark adaptation. The genetic defect has been localized to the arrestin gene, which is responsible for terminating the signaling that triggers cellular response in the rod phototransduction cascade (22). These patients have a good prognosis, with near-normal vision that remains stable (23).
Fundus Albipunctatus
Fundus albipunctatus is another stationary form of night blindness. Patients present with nyctalopia and have essentially normal visual acuity, color vision, and visual fields. This presentation is identical to that of congenital stationary
night blindness and Oguchi’s disease, but fundus albipunctatus is easily differentiated by the presence of multiple white dots scattered throughout the fundus (24) (Fig. 14.13), most likely at the level of the RPE. Patients with fundus albipunctatus have normal-appearing vessels and discs. These patients have a good prognosis, because the vision usually remains normal; however, macular degeneration may develop.
night blindness and Oguchi’s disease, but fundus albipunctatus is easily differentiated by the presence of multiple white dots scattered throughout the fundus (24) (Fig. 14.13), most likely at the level of the RPE. Patients with fundus albipunctatus have normal-appearing vessels and discs. These patients have a good prognosis, because the vision usually remains normal; however, macular degeneration may develop.
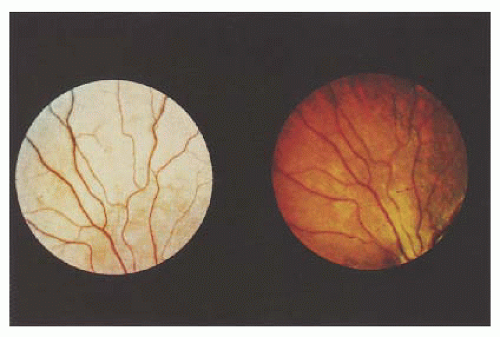 FIGURE 14.12. Oguchi disease and light-adapted retina (left) and darkadapted retina exhibiting the Mizuo phenomenon (right). |
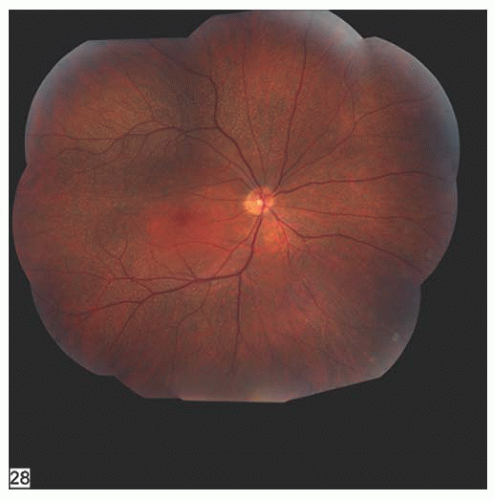 FIGURE 14.13. Patient with fundus albipunctatus with punctate white spots at the level of the RPE throughout the posterior pole, sparing the macula. Note that the disc and retinal vessels are normal. |
This condition is ophthalmoscopically similar to retinitis punctata albescens. However, retinitis punctata albescens is a form of night blindness with progressive retinal degeneration. The discrete uniform white dots in this condition involve the mid-peripheral retina and spare the macula. The autosomal dominant form is associated with a mutation in the human peripherin/RDS gene (25), and the autosomal recessive form is associated with a mutation in the retinaldehyde binding protein gene (RLBP1) (26).
CONGENITAL DEVELOPMENTAL ABNORMALITIES
Aplasia and Hypoplasia of the Macula
Aplasia of the macula is a rare disorder often associated with gross ocular deformities such as microphthalmos, aniridia, coloboma of the optic nerve, monocular myopia, albinism, and medullated nerve fibers. Hypoplasia of the macula, another rare entity, has been suggested as a possible cause of certain forms of amblyopia. In this condition, the central retina does not differentiate completely and is usually arrested at a stage equivalent to 6 to 8 months of intrauterine development. Clinically, this is detected by the lack of the normal perifoveal capillary network, lack of a foveal reflex, absence of the macula lutea pigment, and decreased pigmentation in the foveal pigment epithelium. Visual loss is variable. The cause is uncertain.
Persistent Fetal Vasculature
The most constant feature in PFV, previously known as persistent hyperplastic primary vitreous (PHPV) (27), is a dense, white vitreous band that usually extends from the disc to the fundus periphery or to the lens (Fig. 14.14). It may occur in any meridian but is most common nasally. Limited retinal detachments or other evidence of vitreoretinal traction, such as traction folds, macular pigmentary degeneration, or pigmented demarcation lines, are often associated findings. Prominent uveal processes (Fig. 14.15) and relative microphthalmos are also characteristic of PFV.
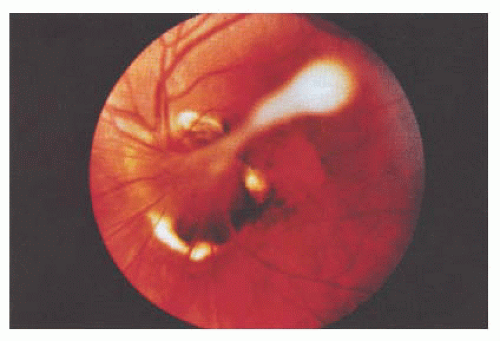 FIGURE 14.14. Persistent hyperplastic vitreous emanating from the disc. |
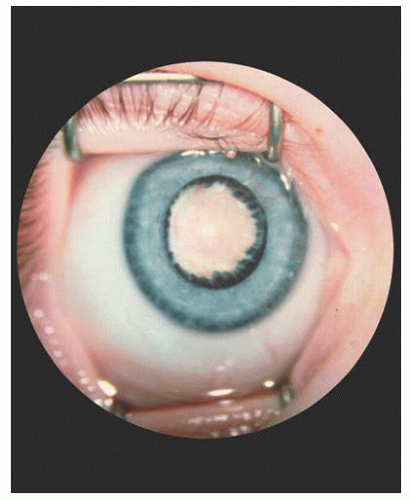 FIGURE 14.15. Elongated ciliary processes and white pupil, characteristic of persistent fetal vasculature (PFV). |
As in other anomalous vascular systems, PFV can vary in degree. The spectrum includes Bergmeister’s papilla, vitreoretinal veils around the disc and macula, vitreous stalks and hyaloid remnants, and retinal folds. Each is related to the other and to congenital abnormalities of the anterior primary vitreous.
The prognosis in PFV is variable depending largely on the severity of the microphthalmia and retinal detachment, if present. Removal of the vitreous opacification and cataract may yield significant visual improvement in selected cases. Aggressive amblyopia therapy is usually necessary.
Myelinated Nerve Fibers
Myelinated nerve fibers occur because of extension of myelination anterior to the lamina cribrosa where it does not belong. The cause is unknown. Myelinated nerve fibers appear during the first year of life, rarely affect visual acuity, and occur predominantly among males; although bilateral cases occur, unilaterality is the rule. The medullated fibers have a feather-like appearance and are usually adjacent to the disc. Sometimes the myelination process is located away from the optic nerve, but unless the macula is affected, vision is preserved (Fig. 14.16). If it is inherited, the mode of transmission is usually autosomal dominant.
Coats’ Disease
Coats’ disease is a nonhereditary abnormality of the retinal vasculature, first described by Coats in 1908 (28). Peripheral retinal telangiectasia, sometimes with a “light bulb” appearance, and secondary exudation are the characteristic findings (Fig. 14.17). In advanced cases, serous detachment of the sensory retina may occur. Discrete dilated vessels and telangiectasia are noted early on fluorescein angiography with marked late leakage. In addition, extensive capillary dropout in areas of peripheral retinal telangiectasia is common. In certain cases, telangiectasia and microaneurysms can occur in the posterior pole and may be associated with exudation posterior to the equator. Macular exudate may also develop in eyes in which the retinal vascular leakage is limited to the periphery. Although the vitreous is usually clear in mild cases, retinal neovascularization and vitreous hemorrhage may occur in advanced cases. Optic nerve involvement in Coats’ disease is rare. In the end stages of Coats’ disease, neovascular glaucoma and phthisis bulbi may develop.
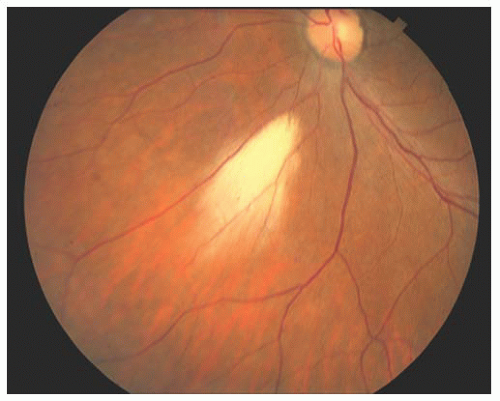 FIGURE 14.16. Myelinated nerve fibers with feather-like appearance away from the optic nerve. |
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


