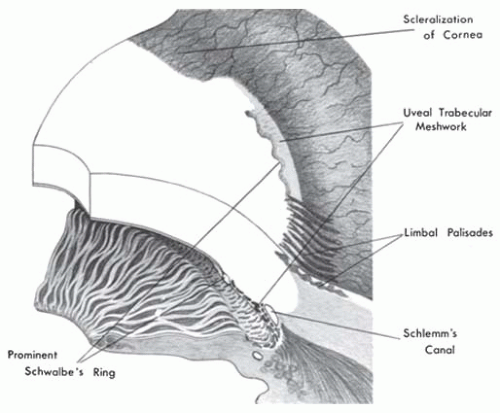
 FIGURE 10.1. Developmental variations in limbal anatomy. This diagrammatic representation of limbal structures illustrates the appearance of each structure on the anterior surface and demonstrates its location in cross section. |
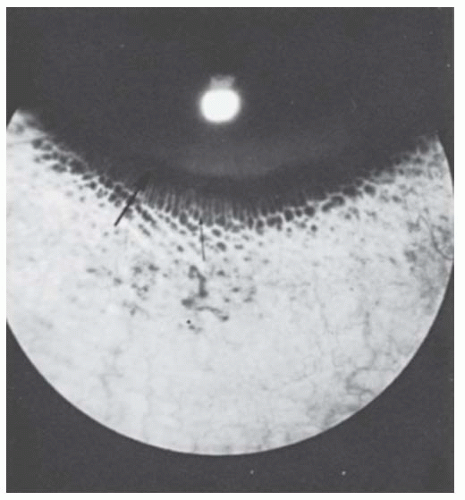 FIGURE 10.2. Limbal palisades of Vogt. This circle of white, fingerlike projections (small arrow) that breaks up the limbal pigment ring (large arrow) results from subepithelial connective tissue papillae pushing up near the surface, interrupting the pigmentation of the basal conjunctival epithelium. |
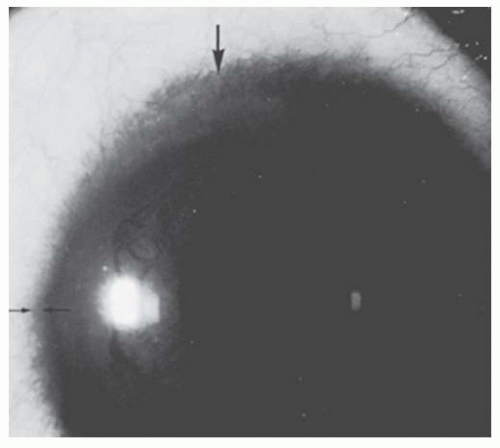 FIGURE 10.3. Scleralization of the cornea. At 12 o’clock position, the sclera and its vessels extend superficially into the cornea, hiding underlying iris and angle details (area between two large arrows). Compare the normal extent of sclera over the limbus at 9 o’clock position (area between two small arrows). |
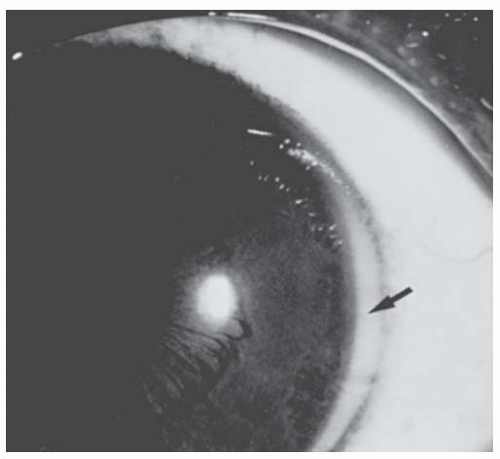 FIGURE 10.4. Prominent uveal trabecular meshwork. The limbus of the eye is demarcated by a pigment ring. Sclera extends up to but not beyond the ring. The light tissue lying central to the pigment ring (arrow) is the uveal trabecular meshwork. |
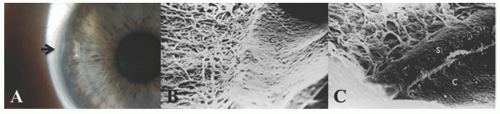 FIGURE 10.5. A: Prominent Schwalbe’s ring (posterior embryotoxon). The limbal pigment ring and prominent uveal trabecular meshwork are present. The distinct white ring demarcating the uveal meshwork centrally is the enlarged, displaced Schwalbe’s ring (arrow), which may be seen in about 10% of normal eyes. B: Scanning electron micrograph of normal Schwalbe’s ring (S), cornea (C), and uveal trabecular meshwork (T) about at this junction. C: Scanning electron micrograph of prominent Schwalbe’s ring (posterior embryotoxon). Endothelium covers both cornea (C) and elevated Schwalbe’s ring (S). (Scanning electron micrographs, courtesy of Morton Smith, MD.) |
megalocornea or anterior megalophthalmos, an X-linked recessive trait that consists of megalocornea, iris and angle abnormalities, and lens subluxation with early cataract formation; and (c) buphthalmos in infantile glaucoma (3). In keratoglobus, the protuberant thin cornea appears enlarged clinically but usually has a normal diameter. There seems to be no entity of “megaloglobus” in which the entire globe is congenitally enlarged with a normal intraocular pressure.
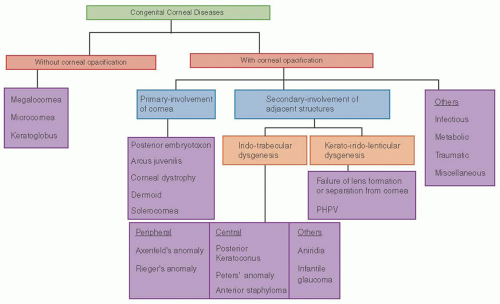 FIGURE 10.6. Flow chart showing clinical classification of congenital corneal diseases. |
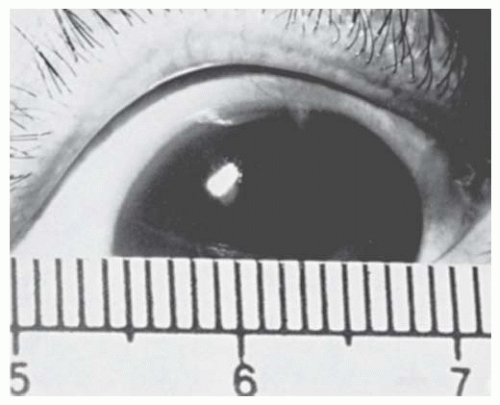 FIGURE 10.7. Anterior megalophthalmos. This cornea measures 15 mm in diameter and has normal thickness and clarity. The disorder is accompanied by transillumination defects in the iris and is associated with lens subluxation and cataract development in the fourth decade. |
glaucoma, spherophakia and/or lens subluxation), which requires lifelong annual examinations for early detection, and cataracts, which often appear in the fourth decade and may require the use of vitrectomy-type instruments during extraction, because the lenses are subluxated or dislocated (6,7). Due to enlarged anterior segment, custom-designed intraocular lenses may be required for lens stability and visual rehabilitation (8).
Table 10.1 DIFFERENTIAL DIAGNOSIS OF ENLARGED CORNEA | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
may produce focal stromal edema (acute hydrops) and heal spontaneously in weeks to months (Fig. 10.8B). Minor blunt trauma to the eye or to the head may rupture the thin cornea and sclera, so the ophthalmologist must counsel the parents of these children to provide a safe environment and protective spectacles or eye guards. Amblyopia is often severe because of the high myopia, a problem diminished by carefully fitted spectacles or contact lenses. A scleral lens may be tried if contact lenses are not successful. In patients with severe thinning and anterior bulging of the cornea, surgical treatment may be contemplated. Given the diffuse corneal thinning to the limbus, surgical repair is problematic at best. A large limbus-to-limbus onlay lamellar keratoplasty or epikeratoplasty to both reinforce the corneal integrity and provide a more normal curvature can be performed. A “tuck-in” lamellar keratoplasty (central lamellar keratoplasty with intrastromal tucking of the peripheral flange) is another technique that can be performed in these patients. If the central cornea is scarred, typically from a previous episode of hydrops, a subsequent central visual penetrating keratoplasty can be performed (9,10,11,12,13,14).
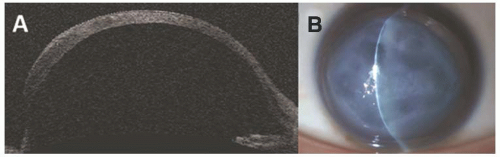 FIGURE 10.8. Keratoglobus A: Anterior segment optical coherence tomography showing ectasia of the entire cornea and midperipheral thinning. B: Slit-lamp photography demonstrating corneal edema due to corneal hydrops in a patient with keratoglobus. |
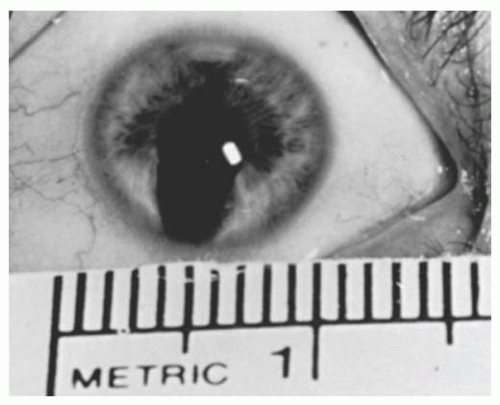 FIGURE 10.9. Microcornea, measuring 9 mm in diameter and accompanied by atypical iris coloboma and congenital cataract. The disorder was inherited as an autosomal dominant trait in this family. |
not seem to be appropriate in the description of abnormalities that occur during embryogenesis and are of neural crest differentiation (22,23,24,25).
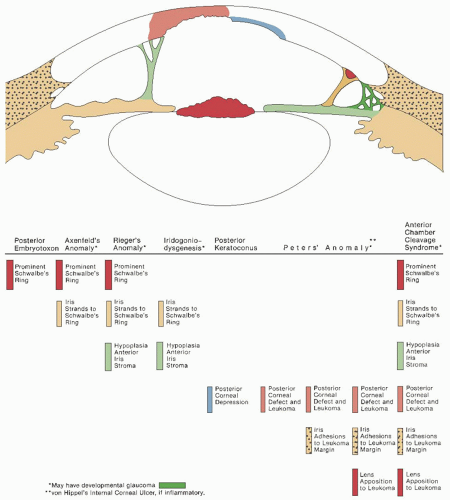 FIGURE 10.10. Composite illustration of the anatomical findings in the anterior chamber cleavage syndrome. The stepladder table demonstrates the spectrum of anatomical combinations and the terms by which they are commonly known. The colored markers in the table indicate the corresponding anatomical component in the illustration. (From Waring GO, Rodrigues M, Laibson PR, et al. Anterior chamber cleavage syndrome: a stepladder classification. Surv Ophthalmol 1975;20:5, with permission.) |
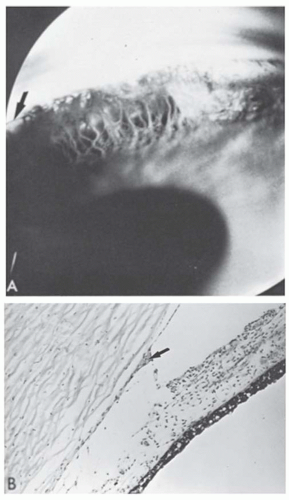 FIGURE 10.11. Axenfeld’s anomaly. A: Gonioscopic view shows the angle recess filled with dense iris processes (persistent mesenchymal tissue) that extend to a prominent Schwalbe’s ring (arrow). This configuration may exist alone or as a part of a variety of iridocorneal dysgeneses (Courtesy of Robinson D. Harley, MD). B: Histologic section showing the prominent centrally displaced Schwalbe’s ring (arrow) with iris processes extending to it and across the angle recess (×64). (Courtesy of Merlyn Rodrigues, MD.) |
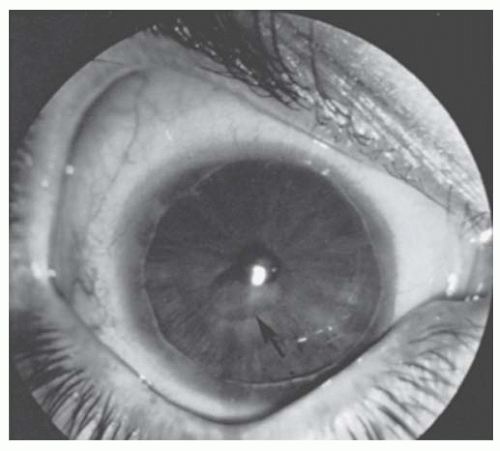 FIGURE 10.12. Rieger’s anomaly with central posterior corneal defect. This right eye of a 23-year-old dwarf demonstrates a prominent Schwalbe’s ring with the iris process extending to it, an atrophic anterior iris stroma, and a central posterior corneal defect (posterior keratoconus) (arrow). Intraocular pressure was normal. The left eye appeared similar. |
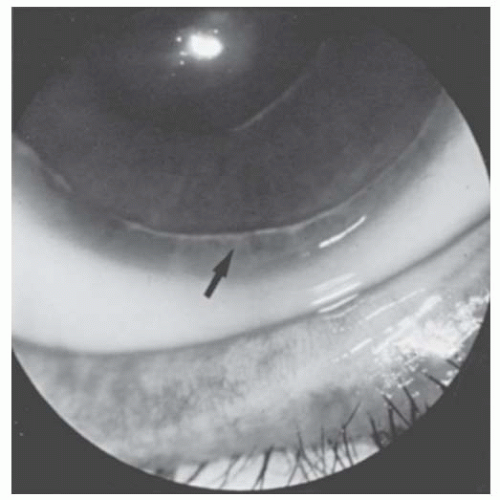 FIGURE 10.13. Rieger’s anomaly. Close-up of 6 o’clock limbus of eye in Figure 10.12. Iris processes (arrow) extend to the irregular prominent Schwalbe’s ring. |
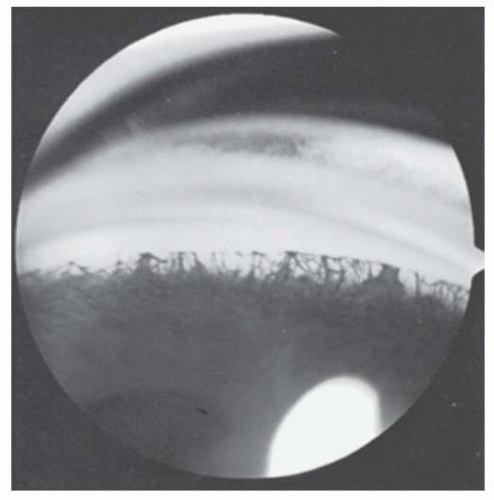 FIGURE 10.14. Angle in Rieger’s anomaly. Gonioscopic appearance of eye in Figures 10.12 and 10.13, showing iris processes extending to the prominent Schwalbe’s ring. |
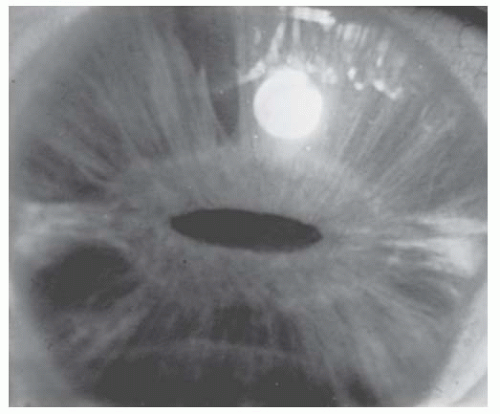 FIGURE 10.15. Rieger’s anomaly. The right eye shows marked hypoplasia of the anterior iris stroma. The deep stroma is thin and fibrillary, revealing the underlying iris epithelium and pupillary sphincter. The pupil is slit-shaped and central. The prominent Schwalbe’s ring is poorly illustrated. (Courtesy of George L. Spaeth, MD.) |
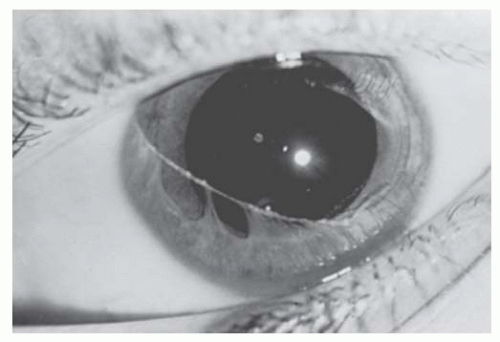 FIGURE 10.16. Rieger’s anomaly. This 10-year-old white girl has a centrally displaced prominent Schwalbe’s ring with iris processes extending to it from the angle recess and the collarette. Anterior iris stroma is absent at 11 o’clock position. The configuration is accentuated by the dilated pupil. Intraocular pressure was normal. No other ocular anomalies existed. (Courtesy of Harold Koller, MD.) |
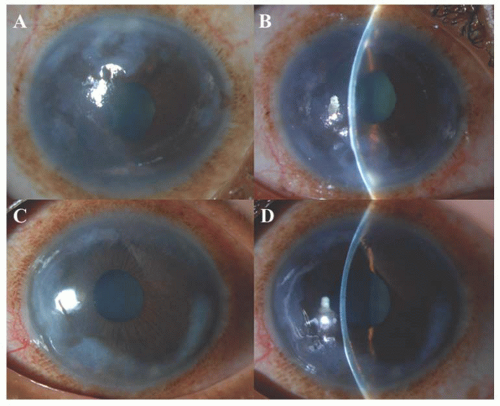 FIGURE 10.17. Rieger’s anomaly. A, B: The right eye demonstrating a peripheral corneal opacity and edema with iris strands anteriorly with peaking of pupil toward 1 o’clock. C, D: The left eye demonstrating peripheral corneal opacity and edema, prominent Schwalbe’s ring nasally with iris processes extending to it, atrophic anterior iris stroma (12-3 o’clock) and also inferiorly on gonioscopy. Intraocular pressure was normal. |
degenerative type (40). Histopathologically, abnormalities include an irregularly thickened epithelial basement membrane, focal disruption of Bowman’s layer, stromal irregularity, and a multilaminar Descemet’s membrane that contains wide-spacing material and focal excrescences (41). Scanning electron microscopy was used to evaluate a cornea with posterior keratoconus, revealing no excrescences of Descemet’s membrane or endothelial tags (42). Given these findings, Al-Hazzaa et al. (42) suggest there may be a subset of posterior keratoconus patients who do not fall into the category of anterior segment dysgenesis abnormalities.
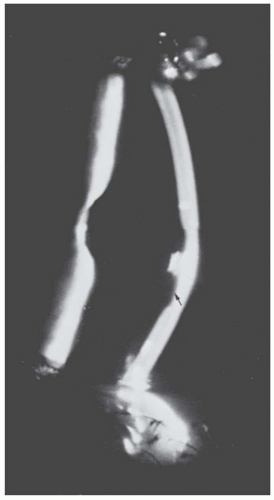 FIGURE 10.18. Posterior keratoconus. Slit-lamp view of the eye in Figure 10.12, showing the depression in the posterior corneal surface (arrow). The cornea overlying it is clear, and no iris processes extend to its margin. |
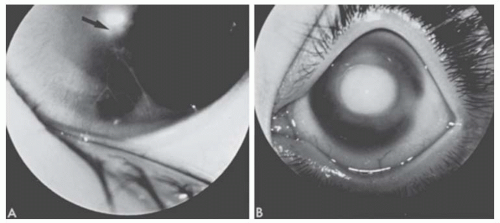 FIGURE 10.19. A: Peters’ anomaly. A mild form showing attenuated iris adhesions to the border of a small corneal opacity (arrow). This was present bilaterally in this 9-month-old white girl. B: Peters’ anomaly. This 10-month-old white girl had bilateral congenital central corneal opacities. During penetrating keratoplasty, iris adhesions were found extending from the pupillary margin to the borders of the opacity. An anterior polar cataract was present. (Courtesy of Harold Koller, MD.) The histopathologic findings are equally varied but usually include thickening or fragmentation of Bowman’s layer, disorganization of stromal architecture, central absence of Descemet’s membrane and endothelium (both of which are present peripherally), and central iridocorneal adhesions (Figs. 10.20 and 10.21C, D). |
 FIGURE 10.20. Peters’ anomaly. Histologic section demonstrates iris adhesions that extend from the collarette to the margin of a central posterior corneal defect. The overlying cornea is edematous. Descemet’s membrane ends abruptly at the margin of the central defect (arrow) (×6). (Courtesy of Robert D’Amico, MD.) |
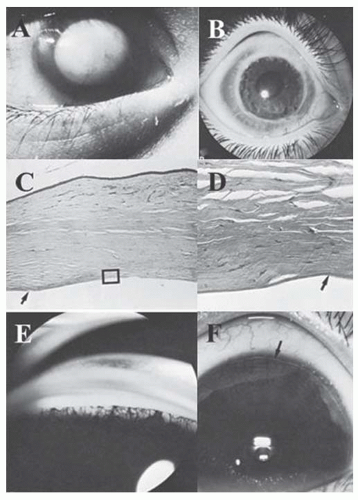 FIGURE 10.21. Anterior chamber cleavage syndrome. A: This 7-month-old boy had bilateral megalocornea (13 mm in diameter), (A-D) a central posterior corneal defect with corneal leukoma in the right eye (Peters’ anomaly), and (E, F) Rieger’s anomaly of the left eye (Courtesy of Turgut Hamdi, MD). B: Keratoplasty for central posterior corneal defect. A penetrating keratoplasty was performed in the right eye at age 22 months, and the graft remained clear for 5 months until graft rejection occurred. No iris processes extended to the corneal leukoma. An anterior polar cataract was discovered postoperatively. C: Central posterior corneal defect with scarring. Histopathologically, the corneal button shows superficial fibrovascular invasion and deep stromal edema. In this area, Bowman’s layer and Descemet’s membrane are absent (box). The margin of the button (left side) shows more normal cornea with edematous stroma. Descemet’s membrane is present in this area (arrow) (Periodic acid-Schiff (PAS) ×25). D: Central posterior corneal defect. The area in the box shows the transition (arrow) from intact Descemet’s membrane peripherally to its replacement by fibrous tissue centrally. Only fragments of endothelium were seen (PAS ×250). E: Megalocornea and Rieger’s anomaly. The left eye of this patient exhibited a 13-mm diameter cornea, a prominent Schwalbe’s ring (arrow) with iris processes extending to it, and a hypoplastic iris stroma. F: Iris processes in Rieger’s anomaly. The angle filled with delicate iris processes and mesenchymal tissue extending up to the prominent Schwalbe’s ring. |
 FIGURE 10.22. Congenital lens-corneal adhesion (Peters’ anomaly). The eye of this newborn demonstrates irregular and thickened corneal epithelium and stroma, central absence of Bowman’s and Descemet’s membranes, a central posterior corneal defect (arrow) with a lens-corneal adhesion, a conical cataractous lens, and malformation of the anterior chamber angles with adhesion of the iris to the cornea. (PAS ×3) (Courtesy of Charles G. Steinmetz, MD.) |
inheritance is also seen. Patients with sporadic aniridia are at risk for WAGR syndrome (Wilms tumor, aniridia, genitourinary abnormalities, mental retardation) and should undergo routine surveillance for kidney disorders. Aniridia is due to mutation at locus 11p13 of the paired box gene 6 (PAX6). It is usually associated with keratopathy, cataract, glaucoma, foveal hypoplasia and strabismus. Dental, musculoskeletal and developmental delays are systemic abnormalities frequently associated with aniridia. Keratopathy is thought to be due to an abnormally differentiated epithelium, abnormal cell adhesion, impaired healing response and limbal stemcell deficiency leading to conjunctivalization of cornea. It begins as vascularized thickening of the cornea at the periphery, which gradually advances centrally. Recurrent corneal erosions lead to subepithelial fibrosis causing corneal opacification. Corneal opacification that occurs due to recurrent erosions is caused by deficiency in matrix metalloproteinase 9 (regulated by PAX6), which is responsible for normal cell remodeling and wound healing. Penetrating keratoplasty for visual rehabilitation is generally unsuccessful because of recurrent surface breakdown. Keratolimbal allograft and Boston keratoprosthesis have proven to be effective for long-term visual rehabilitation (Fig. 10.26) (60,61,62).
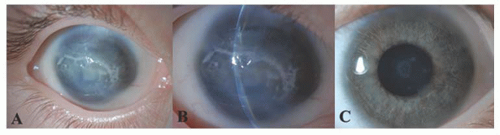 FIGURE 10.23. Peters’ anomaly A, B: The right eye of this child showing central corneal opacity with relatively clear periphery and iris adhesions to the midperiphery and central cornea. C: The left eye of the same patient demonstrating a corneal posterior opacity with an anterior lenticular opacity suggesting a partial dysgenesis. |
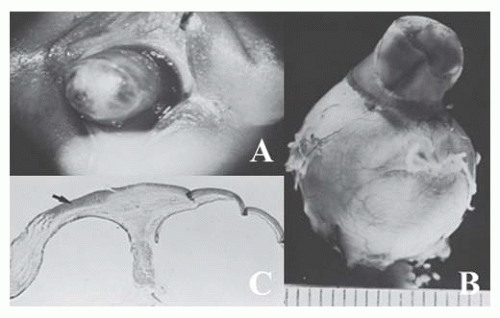 FIGURE 10.24. Congenital corneal staphyloma. A: This 5-day-old infant was born with a flat opaque right cornea. By age 2 days, the cornea had become blue and ectatic, as shown here. The left eye was normal, except for persistent pupillary membrane (Courtesy of Joseph H. Calhoun, MD). B: Gross appearance of the globe. The ectatic area is limited to the cornea (Courtesy of Merlyn Rodrigues, MD). C: Histologic section of globe. Areas of the cornea are thin and ectatic. A superficial corneal abscess from exposure is present (arrow). Bowman’s and Descemet’s membranes are absent. A rudimentary lens is adherent to the central posterior cornea, blending with stromal tissue. Uveal tissue is firmly adherent to the posterior cornea, sweeping down along the lens rudiment (Hematoxylin-eosin ×3). (Courtesy of Merlyn Rodrigues, MD.) |
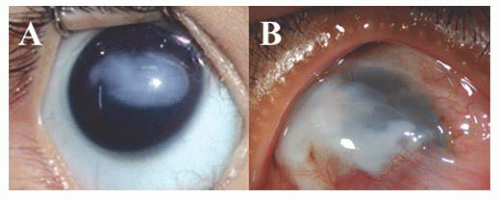 FIGURE 10.25. Congenital corneal keloid. A: 3-month-old boy with corneal keloid and normal intraocular structures, B: Corneal keloid with vascularization in a child associated with Lowe’s syndrome. |
with cornea plana (<38D) and a slightly shallow anterior chamber, which may be a risk factor for the development of secondary glaucoma (66). It should be differentiated from arcus juvenilis, which is devoid of vessels and has a clear lucid interval between the limbus and the corneal opacification.
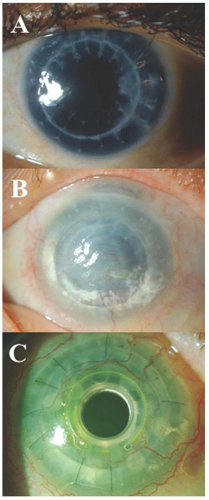 FIGURE 10.26. Aniridia. A: Central clear penetrating keratoplasty in a patient with aniridia. B: Failed penetrating keratoplasty due to aniridiainduced keratopathy. C: Boston keratoprosthesis in a patient with multiple failed grafts. The visual acuity remained 20/400 due to amblyopia and nystagmus. |
Table 10.2 STUMPED: DIFFERENTIAL DIAGNOSIS OF NEONATAL CORNEAL OPACITIES | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
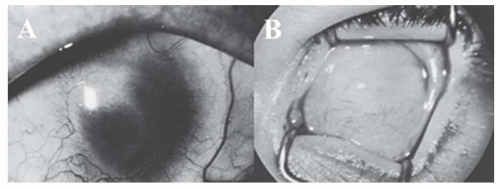 FIGURE 10.27. Sclerocornea. A: Scleral tissue extends in a geographical pattern toward the central cornea. Some clear cornea remains centrally. B: Total replacement of the cornea by sclera. Penetrating keratoplasty was unsuccessful. The iris and lens were grossly malformed. (Courtesy of Joseph Calhoun, MD.) |
rarely its occurrence in families has been reported. The clinical picture is highly variable, however. The masses may be multiple, bilateral, confined to the cornea alone, minutely small, or large enough to obscure the entire cornea (Fig. 10.29). The dermoid extends into the corneal stroma and sclera but seldom occupies the full thickness and only rarely grows into the angle. Hair is not always present on the surface. Dermoids have been classified into three grades: Grade-1, most frequent type and is small (up to 5 mm) and isolated. Grade-2, is much larger and may cover the entire corneal surface and may extend into deeper layers of the cornea. Grade-3, most severe and rare, it replaces the entire anterior segment (74).
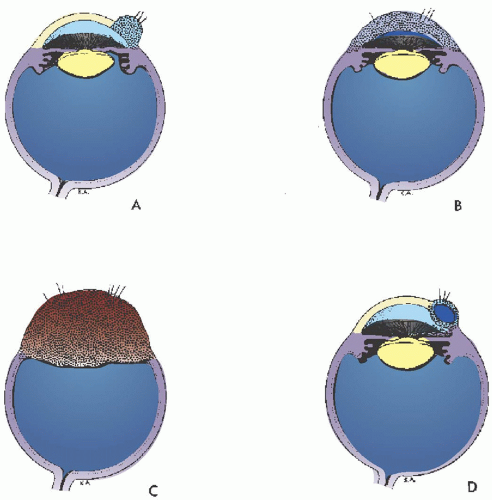 FIGURE 10.28. Corneal dermoids, schematics. A: Limbal dermoid tumor. B: Dermoid tumor replacing the entire cornea. C: Dermoid tumor replacing the entire anterior segment. D: Dermoid cyst of cornea (After Ida Mann). |
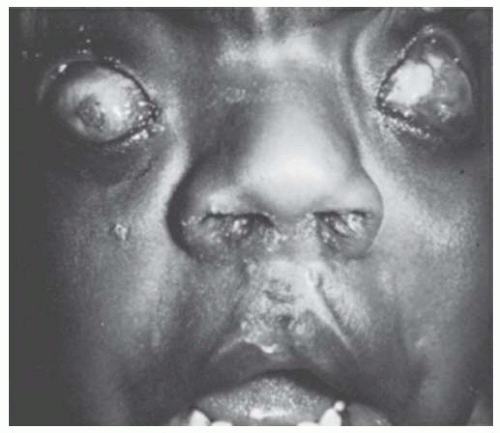 FIGURE 10.29. Bilateral dermoid tumors replacing the entire cornea. This 3-year-old boy was born with masses of vascularized tissue containing surface hair protruding grotesquely between his eyelids. He has had repair of cleft lip and palate. (Courtesy of Robison D. Harley, MD.) |
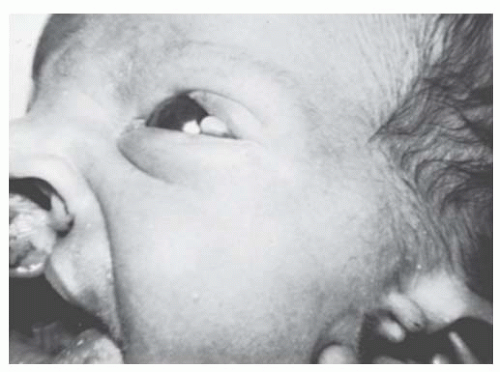 FIGURE 10.30. Goldenhar’s (oculoauriculovertebral dysplasia) syndrome. The limbal dermoid and preauricular skin tags are present, in addition to a cleft lip. (Courtesy of Robison D. Harley, MD.) |
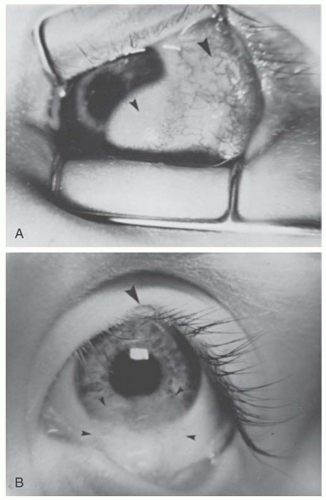 FIGURE 10.31. Goldenhar’s (oculoauriculovertebral dysplasia) syndrome. A: A lipodermoid of the conjunctiva (large arrow) and an epibulbar dermoid of the limbus (small arrow) are present concurrently in about half the cases. B: A coloboma of the upper eyelid at the junction of the middle and inner thirds is present in about one-fourth of cases (large arrow). The limbal dermoid tumor has been excised (small arrows). (Courtesy of Jules Baum, MD.) |
systemic abnormalities, especially cardiovascular, renal, genitourinary, and gastrointestinal defects. Goldenhar syndrome occurs sporadically.
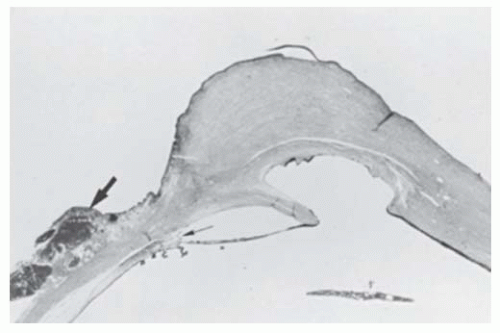 FIGURE 10.32. Corneal dermoid, posterior corneal defect, and Axenfeld’s anomaly. In the right eye, cornea is replaced by a mass of vascularized connective tissue. Ectopic lacrimal gland is present at the limbus (large arrow). In this area the angle is deep and contains a prominent Schwalbe’s ring with iris processes adherent to it (small arrow). A central posterior corneal defect is present. On one side, the iris stretches from the angle to the edge of the defect. Descemet’s membrane is present in this area. On the opposite side, iris lines the corneal defect and posterior cornea; Descemet’s membrane is absent in these areas (Hematoxylin-eosin ×3). |
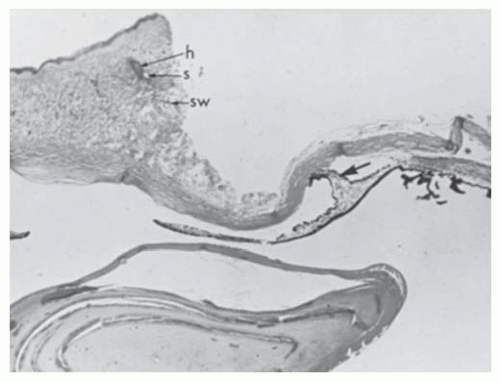 FIGURE 10.33. Corneal dermoid, central posterior corneal defect, and iris-corneal adhesion. The left eye of the patient shown in Figure 10.32. Anterior cornea is replaced by vascularized connective tissue containing hair follicle (h), sebaceous gland (s), and sweat gland (sw). A biopsy has been taken for diagnostic purposes, leaving a defect. Descemet’s membrane is present peripherally but absent centrally. An iris adhesion (arrow) is present centrally. Angle structures are disorganized (Hematoxylin ×4). |
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


