
Diplopia
In this chapter we divide diplopia into several categories: monocular versus binocular and horizontal versus vertical. The evaluation for diplopia is outlined in Figure 10–1.
Is the Diplopia Monocular?
Monocular diplopia can usually be diagnosed by the history alone. Diplopia that is present monocularly remains present despite covering the fellow eye and then disappears when the involved eye is occluded. It may occur unilaterally or bilaterally. The second image is often described as a less clear and partially superimposed “ghost image” or a “halo” on the first image. A pinhole may dramatically reduce the patient’s symptoms. Patients without a clear history of monocular diplopia can be asked to keep a diary of their symptoms with specific instructions to document the details for review at a future visit. A pinhole can be given to patients with suspected monocular diplopia to try at home. This “take home” pinhole can be made in the office out of a business card or a note card using a pen or pencil to make a small-diameter hole. The patient can then try the pinhole at home during the episode of diplopia to test if it resolves the symptoms.
Monocular diplopia usually implies a problem within the eye itself and may respond to refraction, artificial tear trial, or contact lens trial. Table 10–1 lists the ocular causes of monocular diplopia. Monocular diplopia usually does not require any further neuro-ophthalmologic evaluation.
Another less common form of monocular diplopia is cerebral polyopia (Jones, 1999). Cerebral polyopia usually can be distinguished from monocular diplopia due to ocular disease because all of the images are seen with equal clarity, the multiple images do not resolve with a pinhole, and the images are unchanged in appearance whether the patient is viewing binocularly or monocularly. Some patients see only two images, whereas others may see many or even hundreds of images occurring in a grid-like pattern (“entomopia” or “insect eye”) (Lopez, 1993). Some patients experience the polyopia only in certain positions of gaze. Patients with cerebral polyopia often have associated signs of occipital or parieto-occipital region damage, such as homonymous visual field defects, difficulty with visually guided reaching, cerebral achromatopsia or dyschromatopsia, object agnosia, and abnormal visual afterimages. These patients require neuroimaging (e.g., magnetic resonance imaging, MRI), to investigate the etiology of the polyopia. Cerebral infarction is the most common etiology, although cerebral polyopia may also occur with tumors, multiple sclerosis, encephalitis, seizures, and with migraine (Jones, 1999).
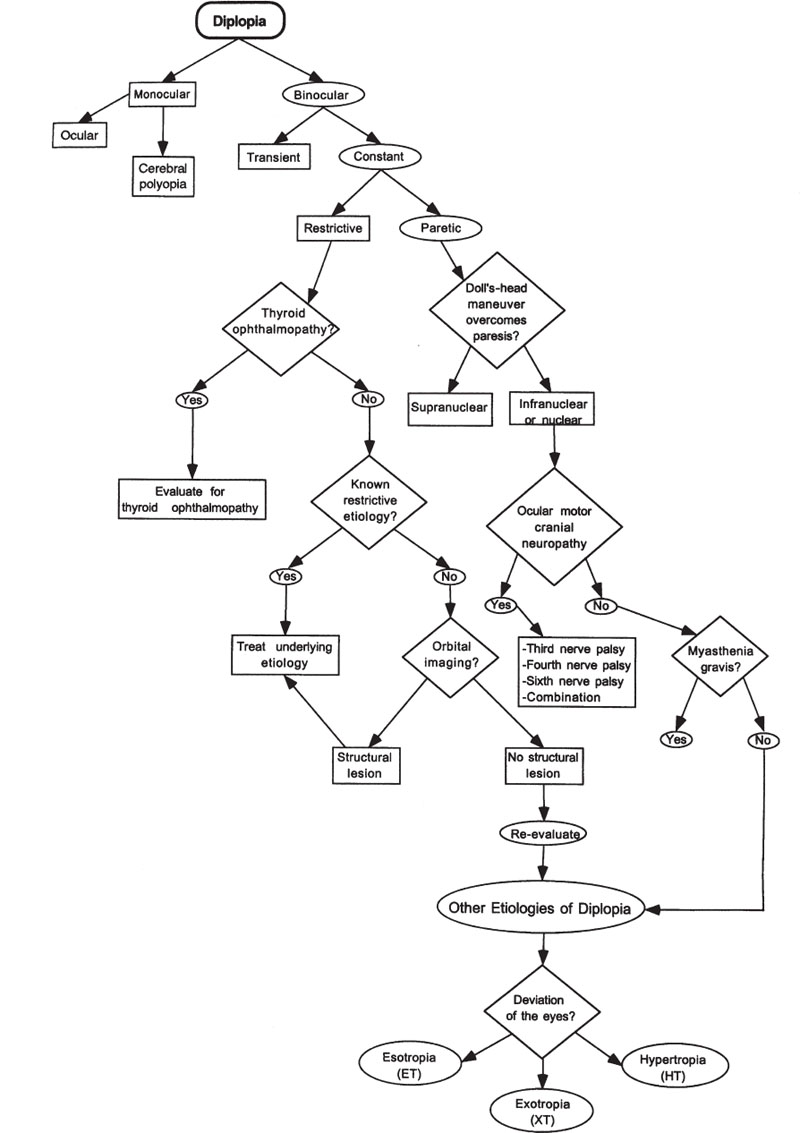
Figure 10–1. Evaluation of diplopia.
Refractive error (Woods, 1996) including astigmatism Poorly fitting contact lens Corneal abnormalities Keratoconus Corneal surface abnormality Tear film disorders including dry eye Refractive surgery Corneal transplant Lid abnormalities (e.g., chalazion, lid position abnormalities) Iris abnormalities (e.g., iridotomy/iridectomy, miotic pupils) Lens abnormalities Cataract Subluxation or dislocation Intraocular lens (e.g., positioning holes, decentered lens) Retinal abnormalities (e.g., epiretinal membrane, scar) |
Is the Diplopia Binocular?
A history of binocular diplopia is associated with ocular misalignment. Identification of specific clinical signs and symptoms may allow identification of specific etiologies for the diplopia.
Is the Diplopia Transient or Persistent?
Diplopia may be noted only in certain fields of gaze (e.g., only on looking down in some patients with fourth nerve palsies) and may fluctuate during the day (e.g., diplopia in thyroid ophthalmopathy may be more apparent in the early morning). Patients with truly intermittent diplopia may be asymptomatic at examination and eye misalignment may be subtle or not demonstrated. Table 10–2 lists the causes of transient diplopia. These etiologies are discussed in more detail in subsequent sections on horizontal and vertical diplopia.
Transient ischemia Transient ocular muscle ischemia (e.g., giant cell arteritis) Vertebrobasilar artery ischemia Decompensation of preexisting phoria Retinal hemifield slide phenomena Myasthenia gravis Muscle or mechanical Thyroid ophthalmopathy Brown’s syndrome Silent sinus syndrome Intermittent phenomena Migraine Neuromyotonia Intermittent or paroxysmal skew deviation Superior oblique myokymia Paroxysmal superior rectus and levator palpebrae spasm Increased intracranial pressure Multiple sclerosis (days to weeks) |
Is This an Ocular Motor Cranial Neuropathy?
Ocular motor cranial nerve palsies are discussed in the chapters on third nerve palsies (Chapter 11), fourth nerve palsies (Chapter 12), and sixth nerve palsies (Chapter 13).
Is There Evidence for a Restrictive Ophthalmoplegia Due to Orbital Disease?
Orbital signs, such as proptosis, chemosis, and injection, should be looked for in patients with diplopia. Forced ductions may reveal a restrictive component to the diplopia. Orbital wall fracture, orbital tumors, orbital inflammatory disease, or trauma may result in a restrictive strabismus. Orbital imaging is indicated in these patients.
Is This Myasthenia Gravis?
The evaluation and management of myasthenia gravis is discussed in Chapter 15. Any patient with painless, pupil-sparing, nonproptotic ophthalmoplegia or diplopia should be evaluated for the possible diagnosis of myasthenia gravis.
Is This Thyroid Eye Disease?
Although transient or persistent diplopia and ophthalmoplegia may occur without other signs of thyroid eye disease, identification of the distinctive signs of thyroid disease as described in Chapter 16 is essential in the evaluation of any patient with diplopia.
Is This a Supranuclear Process?
In a patient with negative forced ductions and no evidence for restrictive ophthalmoplegia, the doll’s-head maneuver (rapid forced head movements horizontally and vertically to activate the vestibulo-ocular reflex) may overcome a supranuclear ophthalmoplegia (see Chapter 14). Failure of the doll’s-head maneuver to overcome the ophthalmoplegia suggests an infranuclear etiology.
Is the Deviation Vertical or Horizontal?
If there are no distinctive or obvious signs to indicate diagnosis of a specific etiology for the diplopia, then the vertical or horizontal nature of the deviation may allow further classification of the problem.
What Are Phorias and Tropias? How Does the Examiner Assess Horizontal Eye Muscle Function?
A phoria is a latent ocular misalignment that is kept in check by fusion. Fusion is the process of merging images from each eye into a single perception. Sensory fusion is the cortical integration of two images, whereas motor fusion represents the corrective movements of the eyes required to maintain eye alignment on the target of regard. Breakdown of fusion due to fatigue, stress, or illness may allow a preexisting phoria to become an intermittent or manifest tropia. The degree of eye deviation may be approximately equal in all directions of gaze (comitant) or less commonly may vary in different positions of gaze (incomitant). Horizontal deviations from decompensation of prior childhood strabismus are typically comitant. Breakdown of acquired deviations, such as an old fourth nerve palsy, may be incomitant.
Ductions (each eye moving separately) and versions (the eyes moving conjugately) must always be assessed. In assessing normal eye excursion, an imaginary vertical line through the lower lacrimal punctum should coincide with a boundary line between the inner third and outer two thirds of cornea. If more cornea is hidden, adduction is excessive; if more cornea is visible and if some sclera is visible, adduction is limited. If abduction is normal, the corneal limbus should touch the outer canthus. If the limbus passes that point and some of the cornea is hidden, abduction is excessive; if some of the sclera remains visible, abduction is limited (von Noorden, 1996).
What Are the Causes of Binocular Horizontal Diplopia (Esotropia and Exotropia)?
Horizontal binocular diplopia is usually due to disease processes affecting the medial and/or lateral rectus muscles, the innervation of these muscles (including ocular motor cranial nerves and neuromuscular junction), or processes affecting fusion or convergence and divergence mechanisms (Brazis, 1999). By definition, patients with horizontal diplopia complain that the two images are side by side. The separation of images may vary or remain unchanged at far or near fixation. For example, the image separation from a left abducens nerve palsy is typically worse at a distance than at close range and worse on left gaze.
Table 10–3 categorizes the causes of binocular horizontal diplopia as either disorders causing esotropia (ET) or disorders causing exotropia (XT). Congenital and childhood strabismus syndromes (Table 10–4) are mentioned but not discussed in depth. For a thorough discussion of childhood strabismus syndromes, the reader is referred to the excellent text of von Noorden (von Noorden, 1996).
What Are the Childhood Strabismus Syndromes Causing Esotropia and Exotropia?
Childhood strabismus syndromes may be confused with acquired causes of ET and XT in adulthood. Most childhood ETs are comitant and present at an early age with “crossed-eyes” or amblyopia. Childhood comitant ETs may be due to hyperopia or impaired accommodation or convergence (Mohney, 2001). Incomitant childhood ETs include A-pattern and V-pattern esodeviations, in which the esodeviation is worse on upward and downward gaze, respectively, retraction syndromes (see below), and mechanical-restrictive esodeviation due to congenital fibrosis of the medial rectus muscle. Some patients with congenital nystagmus are able to decrease the amplitude or frequency of their nystagmus by convergence (nystagmus blockage syndrome) and thus an esotropia develops.
Occasionally, adults with a long-standing, essentially asymptomatic, esophoria may present with diplopia due to “decompensation” (Kushner, 2001). This decompensation of a long-standing esophoria may occur after head trauma, with changing refractive needs, when the patient receives drugs that depress the central nervous system (e.g., alcohol or sedatives), with systemic illnesses, or for unclear reason. History and examination often reveal supportive evidence for a long-standing strabismus, including a history of childhood strabismus or patching, the presence of an old head turn, and horizontal comitance.
Childhood XT is less frequent than childhood ET. The XT may be intermittent or persistent, and sometimes adults with exophoria or intermittent XT may present with diplopia due to the inability to adequately compensate for the eye misalignment (decompensation of exophoria).
Duane’s retraction syndrome is characterized by a narrowing of the palpebral fissure and globe retraction on adduction (Chung, 2000). Three forms have been described (DeRespinis, 1993). In type I, abduction is limited but adduction is normal or only slightly limited. In type II, adduction is impaired but abduction is normal or slightly limited. In type III, both adduction and abduction are impaired. Eye deviation may or may not be present in primary position but if it is present, then ET is usually present in type I and III patients, whereas XT is more frequent in type II patients. Although many patients adopt a head turn to maintain single binocular vision, these patients rarely complain of spontaneous diplopia. They do have diplopia recognition, if specifically asked, and state that they do recognize two images when their eyes are misaligned. In all types, there may be a vertical deviation of the adducting eye with “up-shoots” and “down-shoots.” Duane’s retraction syndrome is predominantly congenital and is thought to be due to anomalous innervation of the lateral rectus muscle by the inferior division of the oculomotor nerve (DeRespinis, 1993). An acquired Duane’s-like syndrome has been described in patients with pontine glioma, with rheumatoid arthritis, following trigeminal rhizotomy, and after removal of an orbital cavernous hemangioma by lateral orbitotomy.
Esotropia Childhood strabismus syndromes (see Table 10–4) Change of angle of preexisting childhood strabismus or loss of suppression scotoma Decompensation of a long-standing esophoria Consecutive esotropia (after strabismus surgery) Optical causes (e.g., optical center change in glasses, over-minus in accommodative esophoria) Sensory esotropia (usually not associated with diplopia) Disorders of muscle and restrictive syndromes Orbital myositis (orbital pseudotumor) Thyroid eye disease Medial orbital wall fracture Postsurgical esotropia Isolated lateral rectus weakness Muscle trauma Progressive external ophthalmoplegia syndromes Anomalous orbital structures, such as extraocular muscles inserting into an abnormal location, fibrous bands, and discrete anomalous muscles (Lueder, 2002) Other orbital disease processes Disorders of the neuromuscular junction (e.g., myasthenia gravis) Disorders of cranial nerves Sixth nerve palsy Ocular neuromyotonia Central disorders Cyclic esotropia Periodic alternating esotropia Divergence insufficiency or paralysis Acute acquired comitant esotropia Spasm of the near reflex Midbrain pseudo-sixth nerve palsy Thalamic esotropia Acquired motor fusion deficiency Hemiheld slide phenomena Exotropia Childhood strabismus syndromes (see Table 10–4) Change of angle of preexisting childhood strabismus or loss of suppression scotoma Decompensation of a long-standing exophoria Consecutive exotropia (after strabismus surgery) Exotropia secondary to vitreous hemorrhage Optical causes Sensory exotropia (often not associated with diplopia) Disorders of the muscle Orbital myositis (orbital pseudotumor) Thyroid eye disease (uncommon) Postsurgical exotropia Isolated medial rectus weakness Muscle trauma Progressive external ophthalmoplegia syndromes Other orbital disease processes Disorders of the neuromuscular junction (e.g., myasthenia gravis) Disorders of cranial nerves Third nerve palsy Ocular neuromyotonia Central disorders Acquired motor fusion deficiency Internuclear ophthalmoplegia (WEMINO syndrome and WEBINO syndrome) and the one-and-a-half syndrome (paralytic pontine exotropia) Vitamin E deficiency (e.g., abetalipoproteinemia) Convergence insufficiency and paralysis Hemifield slide phenomena |
Consecutive esotropia refers to esodeviation that occurs iatrogenically after surgical overcorrection of an exodeviation (patients who are surgically undercorrected may also still be diplopic postoperatively). Consecutive exotropia similarly results from surgical overcorrection of ET or may arise spontaneously in a previously esotropic patient, especially in association with poor vision in the deviating eye (sensory exotropia).
What Are Sensory Esotropia and Sensory Exotropia?
Sensory deviations including ET or XT result from reduced visual acuity in one eye. These patients do not complain of diplopia because of the visual loss. Loss of fusion in cases of visual loss may allow a preexisting phoria to become manifest. Sidikaro and von Noorden reported 121 patients with sensory heterotopias and noted that ET and XT occurred with almost equal frequency when the onset of visual impairment occurred at birth or between birth and age 5 (Sidikaro, 1982). Sensory XT, however, predominates in older children and adults.
Esodeviations Comitant esodeviation Accommodative esotropia Refractive Nonrefractive Hypoaccommodative Partially accommodative esotropia Nonaccommodative esotropia Infantile (onset birth to 6 months) Acquired (includes esotropia with myopia, cyclic esotropia, and some cases of divergence insufficiency) Microtropia Nystagmus blockage syndrome Incomitant esodeviation A- and V-pattern esotropia Duane’s retraction syndrome type I and III Congenital mechanical-restrictive esodeviations (e.g., congenital fibrosis) Secondary esodeviation Sensory esotropia Consecutive esotropia (after strabismus surgery) Exodeviations Primary Duane’s syndrome type II Restrictive—congenital fibrosis Secondary Sensory exotropia Consecutive exotropia (after strabismus surgery) |
Source: Reprinted from von Noorden, 1996, with permission from Elsevier Science.
What Disorders of the Extraocular Muscles Are Associated with Horizontal Diplopia?
Orbital pseudotumor is an idiopathic orbital inflammatory condition characterized by the following features: (1) typically unilateral but may be bilateral; (2) clinical signs of orbital mass effect and inflammation (e.g., proptosis, Chemosis, pain, injection, ophthalmoplegia); (3) orbital imaging shows focal or diffuse inflammatory lesion; (4) histopathology reveals a fibro-inflammatory lesion; and (5) no other identifiable local or systemic causes (Lacey, 1999; Mombaerts, 1996).
When the inflammatory process is confined to one or multiple extraocular muscles, the process is referred to as orbital myositis, although some authors feel that orbital pseudotumor and orbital myositis may be distinct clinicotherapeutic entities (Mombaerts, 1997). Patients present with acute or subacute orbital pain and diplopia. Findings include conjunctival Chemosis and injection, ptosis, and proptosis. Angle-closure glaucoma may rarely occur (Bernardino, 2001). The process may be unilateral or bilateral and usually resolves with corticosteroid therapy (Mombaerts, 1997) or radiation therapy. The illness is often monophasic but recurrent episodes may occur. Characteristics associated with recurrences include male gender, lack of proptosis, eyelid retraction, horizontal extraocular muscle involvement, multiple or bilateral extraocular muscle involvement, muscle tendon sparing on neuroimaging, and lack of response to steroids or nonsteroidal antiinflammatory agents (Mannor, 1997). Orbital myositis may be associated with systemic diseases, such as Crohn’s disease (Squires, 1991), celiac disease, Churg-Strauss syndrome (Takahashi, 2001), systemic lupus erythematosus (Lacey, 1999; Serop, 1994), Whipple’s disease (Orssaud, 1992), rheumatoid arthritis, linear scleroderma (Ramboer, 1997; Serup, 1994; Suttorp-Schulten, 1990), and Wegener’s granulomatosis. Recurrent orbital myositis may occasionally be familial (Maurer, 1999) and orbital myositis may occasionally be paraneoplastic (Harris, 1994).
Neuroimaging reveals enlarged, irregular muscles usually with tendinous insertion involvement (as opposed to tendon sparing in thyroid ophthalmopathy). Intracranial extension of the inflammatory process is rare (De Jesus, 1996). The differential diagnosis of orbital pseudotumor is outlined in Table 10–5.
Orbital polymyositis and giant cell myocarditis is a rare, distinct nosologic entity characterized by progressive, often painful bilateral ophthalmoplegia with thickened extraocular muscles and cardiac arrhythmia often leading to death (Kattah, 1990; Leib, 1994; Stevens, 1996). Pathologically, the extraocular and cardiac muscles showed diffuse mononuclear and giant cell inflammation. Cardiac transplantation may be lifesaving (Leib, 1994).
Biopsy may be required to exclude other diseases, except in pure myositic locations, in which the clinicopathologic picture is rather unique and surgical biopsy may damage the muscle, and in posterior locations, in which the optic nerve may be at risk during surgery (Mombaerts, 1996). Pathologic studies in orbital myositis reveal inflammatory infiltrate composed mainly of small well-differentiated mature lymphocytes, admixed with plasma cells, in a diffuse or multifocal pattern. The muscle fibers are swollen and separated by edema and fibrosis with loss of normal striations and degeneration of muscle fibers (Mombaerts, 1996). Other atypical histopathologic patterns, such as extensive sclerosis, true vasculitis, granulomatous inflammation, and tissue eosinophilia, can be used for subclassification of orbital pseudotumor in general (Mombaerts, 1996). There is no unequivocal correlation between clinicotherapeutic outcome and these atypical findings.
Thyroid eye disease (thyroid orbitopathy, thyroid ophthalmopathy, or Graves’ disease) is a disorder characterized clinically by lid retraction, lid lag in downward gaze, exophthalmos, diplopia (due to extraocular muscle inflammation or fibrosis), potential visual loss due to compressive optic neuropathy or corneal damage, and signs and symptoms of orbital congestion (Bartley, 1994, 1995a,b, 1996a,b). The restrictive extraocular muscle involvement may be confirmed by impaired ocular motility during the forced duction test. The extraocular muscles predominantly affected include the inferior, medial, and superior rectus muscles, and as the process causes muscle tightness or restriction, the diplopia is worse in the direction opposite to that of the involved muscle(s) action. Thus, hypertropia and esotropia are quite common in thyroid eye disease but exotropia is uncommon because lateral rectus muscle is usually not markedly involved. In fact, if a patient with thyroid eye disease is noted to be exotropic, superimposed myasthenia gravis should be considered, as there is an increased risk of myasthenia gravis in patients with thyroid eye disease (Lee 1997; Vargas, 1993). Thyroid eye disease is further discussed in Chapter 16. Thyroid eye disease and orbital myositis may resemble each other clinically. Differential features are outlined in Table 10–6.
Thyroid eye disease (see Table 10–6) Orbital cellulitis (e.g., orbital apex syndrome) Bacterial Fungal Aspergillosis (Hutnik, 1997; Levin, 1996; Slavin, 1991) Mucormycosis (Balch, 1997; Dooley, 1992; Downie, 1993; Johnson, 1999) Bipolaris hawaiiensis (Maskin, 1989) Actinomycosis (Sullivan, 1992) Cysticercosis (Lacey, 1999) Trichinosis (Behrens-Baumann, 1990) Low-flow dural-cavernous sinus fistula Neoplastic Metastatic Breast cancer (false “orbital pseudotumor” presentation) (Goldberg, 1990a,b; Lacey, 1999; Toller, 1998) Lymphoid hyperplasia Non-Hodgkin’s lymphoma and Hodgkin’s disease Sinus histiocytosis with massive lymphadenopathy (Rosai-Dorfman disease) Seminoma (bilateral nonspecific inflammatory or Graves-like orbitopathy not due to direct orbital metastasis) Infiltrative Erdheim-Chester disease (idiopathic infiltration of the heart, lungs, retroperitoneum, bones, and other tissues by xanthomatous histiocytes and Touton giant cells) (Esmaeli, 2001; Shields, 1991; Valmaggia, 1997) Orbital amyloidosis (^eviker, 1997; Lacey, 1999; Murdoch, 1996) Inflammatory Sarcoidosis (Cornblath, 1993; Lacey, 1999; Patel, 1994; Segal, 2000; Takahashi, 2000) Giant cell arteritis (de Heide, 1999) Orbital polymyositis and giant cell myocarditis Systemic inflammatory diseases (Wegener’s granulomatosis, systemic lupus erythematosus) (Woo, 2001) |
The diagnosis of myasthenia gravis (MG) should be considered in all patients with painless ptosis and/or ocular motor weakness without pupillary involvement or proptosis. Weakness and fatigue confined to the extraocular muscles or lids combined with orbicularis oculi paresis is especially suggestive of this diagnosis. MG may cause hypertropia, esotropia, or exotropia, and can mimic many neurogenic conditions including abducens nerve palsies, gaze abnormalities, divergence paresis, and inter-nuclear ophthalmoplegia. Therefore, in any patient with an abnormality of horizontal gaze, MG should at least be considered. MG is discussed further in Chapter 15.
Orbital trauma may result in horizontal diplopia due to a fracture of the medial orbital wall in isolation or accompanied by fracture of the orbital floor or other orbital bones (Eitzen, 1991; Merle, 1998; von Noorden, 1996). Medial rectus muscle incarceration may lead to ET with impaired abduction or XT with impaired adduction. Medial orbital wall injury may occur iatrogenically during endoscopic transnasal sinus surgery (Eitzen, 1991; von Noorden, 1996). Also, medial or lateral orbital surgery (e.g., optic nerve sheath fenestration) may directly injure the medial or lateral rectus muscles, resulting in initial muscle paresis, sometimes followed eventually by scarring and restrictive ET or XT, respectively. Other ocular surgeries (e.g., pterygium surgery, scleral buckle, and glaucoma setons) may also result in horizontal diplopia.
Orbital Myositis | Thyroid Eye Disease |
|---|---|
Males and females equally affected Acute or subacute onset Often severe orbital pain Motility problems early May have limited ductions No lid lag or retraction | Females predominate Gradual onset Painless or “foreign body” sensation Motility problems late Restrictive ductions Lid lag and retraction |
Neuroimaging of orbit | |
Enlarged muscles irregular Tendon spared Often unilateral | Enlarged muscles often smooth Tendon may be involved Often bilateral |
Isolated medial rectus paresis is rare and results in XT, due to unopposed action of the lateral rectus muscle. The XT is worse on gaze to the opposite side and is associated with impaired adduction on the side of the weak muscle. Impaired monocular adduction is more often noted with internuclear ophthalmoplegia than isolated medial rectus palsy due to a partial third nerve palsy. Isolated medial rectus muscle paresis may occur with MG, orbital myositis, muscle trauma, or orbital disease. Lesions of the oculomotor nerve cause medial rectus paresis but not in isolation. Because the neurons controlling the medial rectus muscle probably lie at three different locations within the oculomotor nucleus, it is unlikely that a medial rectus paralysis could be the sole manifestation of a brainstem oculomotor nuclear lesion.
Although isolated lateral rectus paresis is most often due to lesions of the sixth cranial nerve, other processes, including MG, orbital myositis, muscle trauma, and orbital lesions, may impair the muscle directly.
What Disorders of the Cranial Nerves Cause Horizontal Diplopia?
Unilateral sixth cranial nerve injury results in an incomitant esodeviation that is worsened with gaze into the field of the weak lateral rectus muscle. Patients may employ a compensatory face turn in the direction of the paralyzed lateral rectus muscle to limit diplopia. Abduction is often limited on the side of the lesion. With bilateral paralysis, both eyes may be in a position of adduction and the esotropia increases upon looking to the left or right. MG may mimic an isolated sixth nerve palsy, so in some patients with isolated abduction paresis a Tensilon test should be considered, especially if there are signs of fatigability of the muscle paresis or associated ptosis. Sixth cranial nerve palsies are further discussed in Chapter 13.
Lesions of the third cranial nerve may cause an XT because of weakness of the medial rectus muscle with the eye deviating toward the side of the preserved lateral rectus muscle. This XT is usually associated with other signs of third nerve affection, including paresis of eye elevation and depression, ptosis, and pupillary involvement. Third cranial nerve palsies are further discussed in Chapter 11.
Ocular neuromyotonia (ONM) is a rare disorder characterized by episodic (lasting seconds to minutes) horizontal or vertical diplopia, occurring either spontaneously or following sustained (10 to 20 seconds) eccentric gaze (Abdulla, 1999; Barroso, 1993; Chung, 1997; Ezra, 1996b; Frohman, 1995; Fu, 1995; Haupert, 1997; Helmchen, 1992; Morrow, 1996; Newman, 1993; Yee, 1998). Most patients have had prior radiation therapy to the sellar or parasellar region (months to years before onset of the ONM) for tumors, including chordoma, pituitary tumors, craniopharyngioma, chondrosarcoma, rhabdomyosarcoma, thalamic glioma, sinonasal carcinoma, and medulloblastoma. In some cases, however, no responsible structural lesion or history of radiation therapy is noted. Rarely ONM may be due to a compressive lesion, such as an aneurysm (Abdulla, 1999; Ezra, 1996b), dolichoectatic basilar artery (Tilikete, 2000), thyroid eye disease (Chung, 1997), Paget’s disease of bone (Boschi, 1997), or after cavernous sinus thrombosis secondary to mucormycosis (Harrison, 1997). One patient had fourth nerve involvement where spasms of the superior oblique muscle were induced only by alcohol intake (Ezra, 1996b), whereas another developed ONM several years after myelography with thorium dioxide (Thorotrast) (Yee, 1998).
ONM is thought to reflect impaired muscle relaxation due to inappropriate discharges from oculomotor, trochlear, or abducens neurons or axons with unstable cellular membranes. Patients with ONM often benefit from membrane stabilizing agents such as carbamazepine. One patient noted that she could terminate episodes of episodic ocular depression instantly by forcefully directing her gaze upward, and thus stretching the affected muscle might also prove to be an effective way of ending attacks (Safran, 1998). Patients with unexplained transient episodic diplopia should thus be specifically tested for diplopia and ocular misalignment following sustained eccentric gaze.
What Central Disorders Cause Horizontal Diplopia?
Central disorders causing horizontal diplopia include cyclic esotropia, periodic alternating esotropia, divergence insufficiency and paralysis, convergence spasm, convergence insufficiency and paralysis, acquired motor fusion deficiency, internuclear ophthalmoplegia and the one-and-a-half syndrome, vitamin E deficiency, and the hemifield slip phenomenon. Exotropia due to vitreous hemorrhage is included here, as the diplopia may be due to impaired fusional mechanisms. Internuclear ophthalmoplegia, the one-and-a-half syndrome, and the motility disorder associated with vitamin E deficiency (abetalipoproteinemia) may all cause horizontal diplopia (and occasionally vertical diplopia when associated with skew deviation) and are discussed in Chapter 17.
What Is Cyclic Esotropia?
Cyclic esotropia is a rare condition characterized by a regularly recurring ET that often occurs with regular 48-hour cycles (Riordan-Eva, 1993; Tapiero, 1995). There is often a 24-hour period of normal binocular vision followed by a 24-hour period of manifest ET; 72-hour and 96-hour cycles have also been reported. The ET may eventually become constant. Cyclic ET usually appears in young children but may also occur in adults (Riordan-Eva, 1993; Tapiero, 1995). The condition usually starts without precipitant but has been described after strabismus surgery for intermittent XT, after cataract surgery, after traumatic aphakia, after surgical removal of a third ventricular astrocytoma, and in association with optic atrophy or retinal detachment (Riordan-Eva, 1993). The etiology of this condition is unknown, with possible causes being oculomotor nerve hyperactivity (although there are no associated abnormalities of the pupil or lid), abducens nerve dysfunction, strabismus being interrupted by periodic intervals of fusion, or, most likely, a disorder of central mechanisms.
What Is Periodic Alternating Esotropia?
Periodic alternating esotropia (PAE) is a rare cyclic disorder typically associated with periodic alternating nystagmus or periodic alternating gaze (PAG) (Hamed, 1992). While one eye maintains fixation, the other eye undergoes a phase of waxing then waning inward deviation. The cycle is completed by a phase of varying inward deviation in the eye that was initially fixating after a transition period of orthotropia during which fixation changes. This condition is invariably associated with severe brain dysfunction and is especially noted in young children with ataxia or hydrocephalus.
What Constitutes Divergence Insufficiency and Divergence Paralysis?
Weakness of divergence is characterized by intermittent or constant ET at distance with fusion at near (Akman, 1995; Arai, 1990; Friling, 1993; Jacobson, 2000; Lepore, 1999; Lewis, 1996; Schanzer, 1998; von Noorden, 1996). Abduction by duction and version testing is relatively full bilaterally. The angle of strabismus remains unchanged (comitant) or may be decreased on gaze to either side. Fusional divergence is reduced or absent. Fusional divergence is measured by placing prisms of progressively larger strength base-in over one eye while the subject is fixating at distance and near and noting when the fixation image appears double (break point). Patients with divergence weakness should also demonstrate normal speed and amplitude of horizontal saccades (Leigh, 1999).
When ET at distance due to divergence impairment occurs in an otherwise healthy individual, it is referred to as “divergence insufficiency” or “primary divergence insufficiency,” whereas when it occurs associated with neurologic disease it is called “divergence paralysis” or “secondary divergence insufficieny.” Divergence insufficiency (primary) is usually observed in young adults, is self-limited, and may be treated with base-out prisms or occasionally surgery (Akman, 1995; Arai, 1990; Friling, 1993; Jacobson, 2000; Lewis, 1996; Schanzer, 1998; von Noorden, 1996). In one study, 95% of patients with primary divergence insufficiency were older than 50 years and symptoms resolved in 40% of patients after a median of 5 months (Jacobson, 2000). Divergence paralysis (secondary divergence insufficiency) is usually noted with brainstem disease. It has been reported with multiple sclerosis, intracranial masses (e.g., pontomedullary glioma), brainstem hemorrhage or infarction, head trauma, increased intracranial pressure (e.g., pseudotumor cerebri, neurobrucellosis, frontal lobe glioblastoma), the spontaneous intracranial hypotension syndrome, cerebellar lesions, cranio-cervical junction abnormalities (e.g., Chiari malformation), hydrocephalus, meningitis, encephalitis, syphilis, clivus lymphoma, acute lymphoblastic leukemia, chronic lymphocytic leukemia, diazepam ingestion, giant cell arteritis, Fisher’s syndrome, Wernicke’s encephalopathy, Parkinson’s disease, Machado-Joseph disease, progressive supranuclear palsy, and after lumbar puncture or epidural block (Akman, 1995; Arai, 1990; Brown, 1999; Friling, 1993; Horton, 1994; Jacobson, 2000; Lepore, 1999; Lewis, 1996; Mokri, 1997; Ohyagi, 2000; Schanzer, 1998; Tekeli, 1999; Versino, 1996). Abducens nerve palsy may also cause esotropia that is worse at a distance than near, and indeed some authors believe that divergence paralysis does not exist and that all such cases actually represent bilateral abducens nerve palsies. However, three findings occur with divergence paralysis but not with bilateral sixth nerve palsies: (1) fusional divergence is reduced or absent, (2) the esotropia not only remains unchanged during horizontal gaze but may even decrease, and (3) saccadic velocities are normal.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


