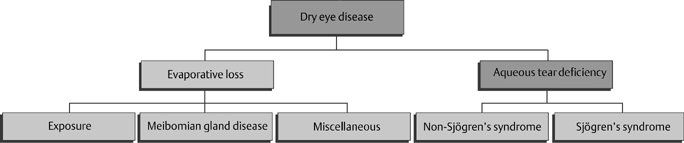
13 Charles D. Reilly and Mark J. Mannis The patient with severe dry eye poses a challenge in both evaluation and management. In this chapter, we will cover a brief review of the classification of the dry eye, the clinical evaluation of the patient with severe dry eye, a review of the causes of severe dry eye, and approaches to treatment. The surgical management of these patients is covered in Chapter 14. To understand severe dry eye, it is first important to understand the classification of dry eye. The National Eye Institute (NEI)/Industry Workshop Classification Scheme for Dry Eye developed in 1995 divides dry eye disorders into those related to deficient aqueous production and those related to evaporative loss (Fig. 13-1).1 The classification further divides each of these main categories into subcategories. Aqueous deficiency is divided into Sjögren’s syndrome (primary and secondary) and non-Sjögren’s syndrome (lacrimal gland disease, loss of reflex tearing, and lacrimal gland duct obstruction). Evaporative loss is divided into exposure, meibomian gland disease, and miscellaneous abnormalities (e.g., blink abnormalities, contact lens problems). This classification is a useful starting point for the understanding of dry eye disease. In the past 5 years our understanding of basic pathophysiology has incorporated the role of inflammation and inflammatory mediators in the pathogenesis of dry eye also.2–6 This classification is useful in the clinical evaluation of patients with dry eye, dividing patients into those with tear deficiency and those with poor tear function, even though there may be significant overlap. The diagnosis of severe dry eye disease is based on clinical findings. There is limited consensus as to what constitutes severity. There are, however, certain criteria the clinician can use to determine severity using subjective or objective criteria. Correlation between a patient’s subjective complaints and objective findings in dry eye is generally poor.7 The Ocular Surface Disease Index (Fig. 13-2) is a subjective measurement tool. This index has been correlated with objective findings and has been validated with other ocular surface disease questionnaires, including the National Eye Institute Visual Function Questionnaire (NEI VFQ-25) and the McMonnies Dry Eye Questionnaire.8 Figure 13-1 Classification scheme of dry eye. (Adapted from Lemp MA. Report of the National Eye Institute/Industry Workshop on Clinical Trials in Dry Eyes. CLAO J 1995; 21:221–232.) Objective findings are also important. Several tests have been established to aid the clinician in determining the severity of dry eye. Schirmer’s tests can be used. Generally, wetting of 5 mm or less (Schirmer’s 1 test) is considered abnormal, and the less wetting the more severe the dry eye disease. Other objective measures employed clinically include tear breakup time (BUT), tear meniscus height, vital dye staining of the ocular surface (Table 13-1), evaluation of the lid margin for signs of inflammation and meibomian gland dysfunction (MGD), and the phenol red test.9 Additional tests not routinely performed include tear film osmolarity, tear lysozyme, and tear lactoferrin.9 These studies and others can be helpful in diagnosing and following the clinical course of patients with dry eye disease. Other chapters in this textbook provide detail concerning each of these tools.
Diagnosis and Treatment of Severe Dry Eye Disease
Key Points
♦ Classification of Dry Eye
Diagnosis of Severe Dry Eye Disease
| Dye | Stains What | Toxicity |
| Fluorescein | Precorneal tear film and disrupted cell | None |
| Rose bengal | Dead and degenerating cells, healthy cells | Direct toxicity to cells, virus, bacteria, and protozoa |
| Lissamine green | Dead and degenerating cells | No toxicity to epithelial cells |
Adapted from Kim J. The use of vital dyes in corneal disease. Curr Opin Ophthalmol 2000;11:241–247.

Evaluation of the precorneal tear film and staining pattern can assist in determining the cause of the dry eye. There are well-recognized staining patterns for specific ocular conditions, and there have been attempts to create grading criteria.1,10,11 The van Bijsterveld grading scale rates the intensity of rose bengal staining on a scale of 0 to 3 in three areas on the eye (cornea, nasal conjunctiva, and temporal conjunctiva).10 The maximum score is 9. Lemp’s grading system incorporates five different corneal zones and includes conjunctival zones.1 These scales help quantify the staining seen.
The Oxford Grading Scheme uses a picture scale (Fig. 13-3) to rate ocular staining with fluorescein, rose bengal, or lissamine green stain. There are six panels with representative pictures of staining. The number of punctate erosions increases by 1 log unit between A and B then by 0.5 log units from B to E. Staining greater than panel E is considered severe.11
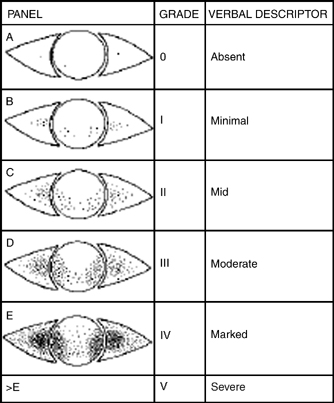
Figure 13-3 Oxford Grading Scheme for dry eye disease. (From Bron AJ, Evans VE, Smith JA. Grading of corneal and conjunctival staining in the context of other dry eye tests. Cornea 2003;22:640–650. Used with permission.)
♦ Causes of Severe Dry Eye Disease
Once a diagnosis of severe dry eye is made, it is important to determine the cause to help guide appropriate therapy.
Congenital Alacrima
Congenital alacrima is characterized by deficient lacrima-tion presenting shortly after birth. This disorder is typically autosomal dominant, but autosomal recessive forms have been reported.12–15 Modino and Brown described congenital alacrima in a family of five persons over four generations showing decreased lacrimation from infancy. Clinical testing and histopathology of the lacrimal gland demonstrates hypoplasia of the lacrimal glands.15 Imaging may demonstrate smaller than normal lacrimal glands.16 Alacrima may also occur in anhidrotic ectodermal dysplasia and dysautonomia and in association with ocular and adnexal abnormalities such as Allgrove’s syndrome, which includes alacrima, achalasia, and adrenocortical insufficiency and Pierre Robin syndrome.17–19 This diagnosis should be suspected in any patient with severe ocular surface dryness at an early age with or without a family history of this disorder.
Riley-Day Syndrome
Riley-Day syndrome, also known as familial dysautonomia and hereditary sensory and autonomic neuropathy type III, is a generalized disorder of the autonomic nervous system. Key features include alacrima, absence of lingual fungiform papilla, indifference to pain and temperature, and vasomotor instability. It is an autosomal recessive disorder that occurs almost exclusively in persons of Ashkenazi Jewish descent.20–22
The clinical diagnosis of Riley-Day syndrome is based on the presence of five signs: lack of flare after intradermal injection of histamine, absence of fungiform papillae on the tongue, miosis of the pupil with the instillation of metha-choline chloride (2.5%), absent deep tendon reflexes, and diminished tear flow.23 Neuropathological findings in sural nerve biopsies may be the best diagnostic criterion to differentiate Riley-Day syndrome from other congenital sensory neuropathies.23,24 The clinician should suspect this syndrome if a child of Eastern European Jewish ethnicity has a history of breech delivery with meconium staining, failure to thrive secondary to poor sucking, hypotonia, or hypothermia. The diagnosis can be confirmed by inspection of the tongue for fungiform papillae, detection of diminished deep tendon reflexes, and performance of intradermal histamine test and ocular pilocarpine tests.25 Prenatal diagnosis is now possible. Eng et al26 used CA-repeat polymorphisms located in the 9q31–q33 region for prenatal diagnosis of Riley-Day syndrome. Six fetal diagnoses were predicted with more than 98% accuracy, whereas two with recombinations were predicted with at least 88 and 92% accuracy.
Anhidrotic Ectodermal Dysplasia
There are more than 150 clinically distinct hereditary syndromes in which ectodermal dysplasia is present. Most are very rare and include defects in ectodermal structures, including hair, skin, nails, and teeth.27,28 Using linkage analysis, anhidrotic ectodermal dysplasia has now been proved to be X-linked.
Saksena and Bixler studied 13 families and described the characteristic facies of patients who suffer from anhidrotic ectodermal dysplasia. Features include a pronounced forehead, small cranial length, small palatal depth, and depressed nasal root and bridge.27,29 There are multiple mutations in the EDA gene responsible for the phenotype,30 including 12 missense, one nonsense, and four deletion mutations.27,31 As a historical note, anhidrotic ectodermal dysplasia was first described by Darwin in 1875. His description of the “toothless men of Sind,” refers to the disorder. He wrote:
I may give an analogous case, communicated to me by Mr. W. Wedderburn, of a Hindoo family in Scinde, in which ten men, in the course of four generations, were furnished, in both jaws taken together, with only four small and weak incisor teeth and with eight posterior molars. The men thus affected have very little hair on the body, and become bald early in life. They also suffer much during hot weather from excessive dryness of the skin. It is remarkable that no instance has occurred of a daughter being affected…though the daughters in the above family are never affected, they transmit the tendency to their sons: and no case has occurred of a son transmitting it to his sons. The affection thus appears only in alternate generations, or after long intervals.27,31
Given these abnormalities, it is unlikely a patient would present to ophthalmology with the diagnosis in question; however, the diagnosis should be suspected in young male patients with severe dry eye and syndromic facies.
Vitamin A Deficiency
Vitamin A deficiency leads to a loss of mucus-secreting goblet cells and eventually to squamous cell metaplasia of the conjunctival epithelial cells. Conjunctival xerosis is typically found on the temporal, interpalpebral, bulbar conjunctiva and appears as thickened dry granular patches that stain with rose bengal.32,33 Bitot’s spots are triangular, perilimbal, gray plaques of keratinized conjunctival debris overlying an area of conjunctival xerosis. These spots resolve with vitamin A intake.33,34
The corneal manifestations of xerophthalmia begin with an unstable tear film and lead to loss of corneal luster and punctate epithelial keratopathy. The keratopathy progresses to epithelial defects, stromal edema, and keratinization. Treatment can reverse these changes. If left untreated, corneal ulcers develop. Such ulcers are typically small and nasal with sharp borders.33
Keratomalacia is a full-thickness liquefactive necrosis of the cornea. Clinically, lesions are sharply demarcated with an opaque grayish yellow appearance. The diagnosis of xerophthalmia is clinical, requiring a high degree of suspicion. Blood tests are available but may require specialized laboratories. Serum vitamin A can be measured directly, and deficiency is a plasma level of 35 μmol/dL or less.32 Conjunctival impression cytology may also be useful for detecting preclinical xerophthalmia. Squamous metaplasia is diagnosed by the presence of irregular, enlarged, and keratinized epithelial cells with loss of goblet cells.35,36
Systemic treatment is required to replenish vitamin A stores. The oral dosage regimen is 200,000 IU of vitamin A in oil followed the next day with an additional dose of 200,000 IU.32 Children who also suffer from severe protein deficiency should receive an additional oral dose every 2 weeks until their protein status improves.32 If patients have severe corneal disease or malabsorption, the customary dose is 100,000 IU of water-miscible vitamin A intramuscularly.37 Topical therapy is used to treat or prevent secondary bacterial infection in the xerophthalmic ulcers. Once systemic treatment is initiated, there is a delay of several days before an effect on ocular tissues occurs. Therefore, topical retinoic acid (0.1%) can be used to bridge that time gap and promote healing.38 Topical retinoic acid can also lead to a more dense, vascularized scar, however, so its use in ulcers in the visual axis requires caution.
Stevens-Johnson Syndrome
Stevens-Johnson syndrome (erythema multiforme major) is an acute, self-limited, inflammatory condition caused by an idiosyncratic reaction to drugs or infection.39,40 It is a severe form of ocular surface disease with a poor visual prognosis.40–43 Late clinical findings include symblepharon, corneal opacification with conjunctivalization, and adhesive occlusion of the lacrimal puncta.44
Figure 13-4 demonstrates typical findings in Stevens-Johnson syndrome, including destruction of the lid architecture and pseudopterygium formation. Tear supplementation with nonpreserved artificial tears is important for the management of the ocular surface disease in Stevens-Johnson syndrome. Drops made of autologous serum may also be useful, providing both wetting and essential tear components, such as epidermal growth factor and vitamin A.45–48 In addition to tear supplementation, anti-inflammatory medications may provide some benefit. Topical corticosteroids and topical cyclosporine have been used with some efficacy.49,50 There is also evidence that punctual occlusion can promote healing by causing retention of tears and essential tear components.41 The administration of systemic immunosuppressive agents is also important. Systemic corticosteroids, thalidomide, dapsone, plasmapheresis, intravenous immunoglobulin, and mycophenolate mofetil have all been reported to be beneficial in treating the systemic manifestations of Stevens-Johnson syndrome.51–55
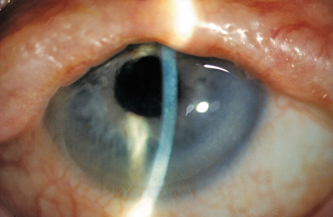
Figure 13-4 Stevens-Johnson syndrome: There is destruction of the lid architecture with vascular ingrowth into the inferior cornea. Note also the pseudopterygium nasal.
Graft-versus-Host Disease
Graft-versus-host disease (GVHD) is a major cause of morbidity and mortality in patients undergoing allogeneic stem cell transplant. There are two major forms of GVHD, acute and chronic. Acute GVHD presents as hepatitis, enteritis, and dermatitis within 100 days of allogeneic stem cell transplant. Chronic GVHD generally develops after day 100 of allogeneic stem cell transplant and has a more varied presentation.56 Chronic GVHD can present 3 to 14 months after hematological stem cell transplant and affects 40 to 60% of matched, unrelated donor recipients and 20% of matched sibling transplants.56
The eye is one of the sites most commonly involved.56,57 Dry eye associated with chronic GVHD is one of the major late complications after allogeneic stem cell transplantation. It affects patient quality of life and can lead to blindness.56–59 Dry eye disease associated with chronic GVHD is the most frequent ocular complication after stem cell transplantation (40 to 60%), followed by MGD (48%) and retinal hemorrhage (3.5 to 20%).56,58–60 Severe dry eye disease resembling Sjögren’s syndrome progresses rapidly after the onset of symptoms in most patients.59 MGD is frequently associated with severe dry eye related to chronic GVHD.59 Therefore, the presence of MGD is helpful for the diagnosis of dry eye associated with chronic GVHD. There is a higher risk for the development of ocular involvement in chronic GVHD in patients who undergo peripheral blood stem cell transplantation than in patients who undergo bone marrow stem cell transplantation.61
Chronic GVHD is a systemic, inflammatory alloimmune response to the recipient cells by donor lymphocytes that resembles an autoimmune disorder.56
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


