Craniofacial
Alex V. Levin
Thomas W. Wilson
J. Raymond Buncic
The craniofacial disorders are characterized by malformation of the cranial vault and/or facial bones. Premature fusion of one or more sutures, known as craniosynostosis, results in myriad congenital syndromes that are distinguished not only by the involved sutures and appearance of the head and face, but also by the presence and absence of other systemic findings. The genes involved in craniofacial development may also be involved in the formation of other body parts. Clefting abnormalities are a group of disorders that are the result of incomplete closure of the face and branchial clefts as defined by the Tessier clefting system. One must also distinguish between primary craniofacial malformations and the secondary deformations such as positional plagiocephaly.
Craniofacial disorders often have ophthalmic manifestations. A multispecialty team approach is required for the care and treatment of these patients. Significant vision loss can be observed due to primary coexisting malformations such as optic nerve hypoplasia, secondary effects of the disorder such as corneal exposure due to exorbitism or strabismic amblyopia, and complications of treatment such as hemorrhagic conjunctival prolapse following a bicoronal flap for surgical intervention.
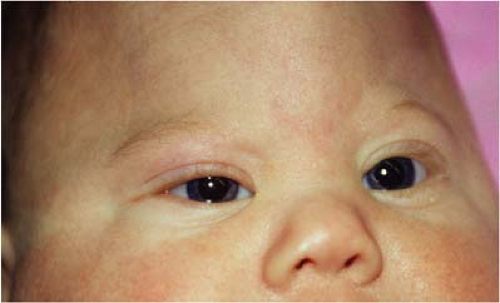 Figure 14.1 Metopic Synostosis Metopic craniosynostosis (trigonocephaly) is due to premature closure of the metopic suture of the skull. This causes a wedge-shaped malformation of the frontal bone, which leads to hypotelorism and pseudoesotropia. Increased intracranial pressure is rare. This is usually an isolated anomaly that does not affect cognition but may result in significant departure from a normal appearance. |
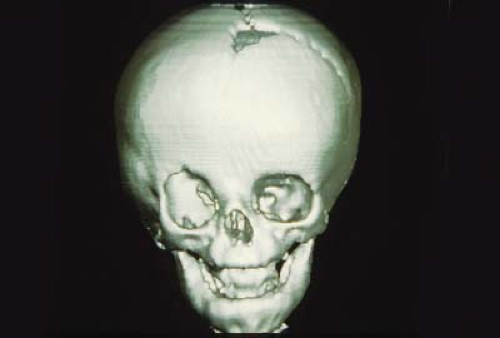 Figure 14.2 Coronal Synostosis Coronal craniosynostosis (anterior plagiocephaly) is due to premature closure of the coronal suture. This three-dimensional reconstruction of the skull shows premature closure of the right coronal suture. Note the posterior displacement of the superior and superolateral orbital rim, sometimes referred to as the “owl eye” or “harlequin orbit.” Bilateral coronal synostosis may also. Although often associated with other abnormalities of the skull or body, unilateral or bilateral coronal synostosis may also occur as an autosomal dominant condition due to mutations in the fibroblast growth factor receptor gene FGFR2 at 10q26. Elevated intracranial pressure may be present, particularly in bilateral cases. |
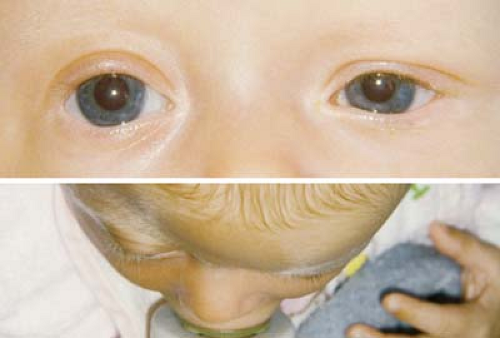 Figure 14.3 Coronal Synostosis Due to the superior and superolateral orbital rim recession, patients with coronal synostosis (anterior plagiocephaly) appear to have a recessed forehead on the affected side and pseudoproptosis. Misdiagnosis as buphthalmos (Chapter 10: Glaucoma, Fig. 10.1), contralateral hemifacial atrophy, or contralateral ptosis is not uncommon. The view from above (bottom image) is very useful in evaluating patients with craniofacial disorders and in this case immediately reveals the diagnosis. |
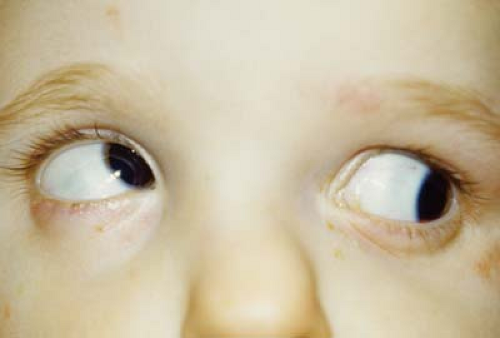 Figure 14.4 Coronal Synostosis Approximately 50% of patients with anterior plagiocephaly will have a vertical and/or horizontal strabismus with limited eye movements. This child has limited supraduction in abduction and eye movements consistent with bilateral inferior oblique overaction. This abnormal pattern may be associated with absent or malpositioned eye muscles, particularly in bilateral cases. Astigmatism is more common on the involved side(s). |
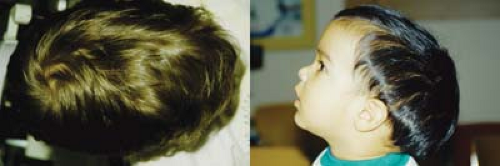 Figure 14.5 Sagittal Synostosis Sagittal synostosis (scaphocephaly) is due to premature closure of the sagittal suture. The skull is elongated in an anterior posterior direction. Often, there will be a palpable ridge along the fused suture and the anterior and posterior fontanelles will be completely or partially closed. Increased intracranial pressure is not uncommon and papilledema is the major ocular sign. Until cranial vault reshaping is performed, periodic ocular examination is suggested. |
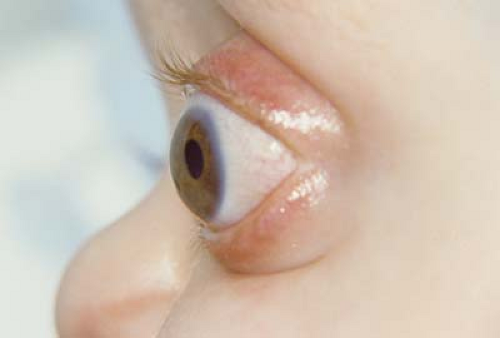 Figure 14.6 Exorbitism In the multiple craniosynostosis syndromes the orbit is relatively shallow, leading to prominence of the globe. This is different than proptosis in which there is something (e.g., tumor) pushing the globe forward. Exorbitism can result in spontaneous and recurrent subluxation of the globe. Severe exorbitism can result in the lids being positioned behind the equator. Exposure keratopathy is a particular concern. Although subluxation may be treated by firm gentle pressure to reposit the globe in the orbit, craniofacial surgery is required for a more definitive result. |
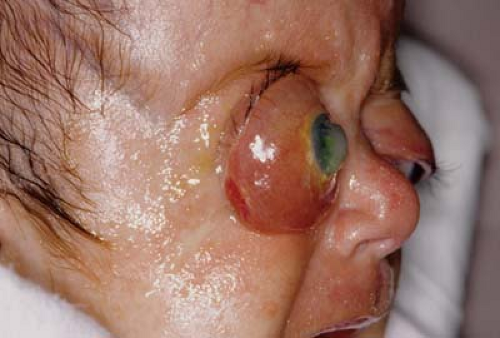 Figure 14.7 Complicated Exorbitism Severe exorbitism can lead to conjunctival edema and corneal exposure, particularly if the eye becomes trapped anterior to the orbital rim, as seen here. Compromise to the optic nerve may occur. If the eye cannot be manually reposited within the orbit, treatment of this patient acutely would involve aggressive lubrication of the conjunctiva and cornea. Tarsorrhaphy is very difficult unless the eye can be placed back in the orbit. This patient required urgent advancement of the orbital bones. |
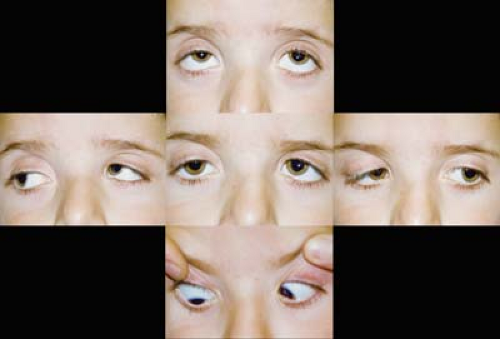 Figure 14.8 Multiple Craniosynostosis Strabismus The most common strabismus is a V-pattern exotropia with hypertropian adduction. Extraocular muscle anomalies are common and lead to strabismus. Anomalies include absent or malpositioned muscles with atypical insertion sites. Satisfactory surgical management of this strabismus is difficult. The elevation in adduction and V pattern might suggest an overaction of the inferior oblique muscles (Chapter 1: Strabismus, Figs. 1.28, 1.31, and 1.38), but the mechanism is most likely more complex. This pattern has been observed even in children with multiple craniosynostosis, exorbitism, and absent inferior oblique muscles. |
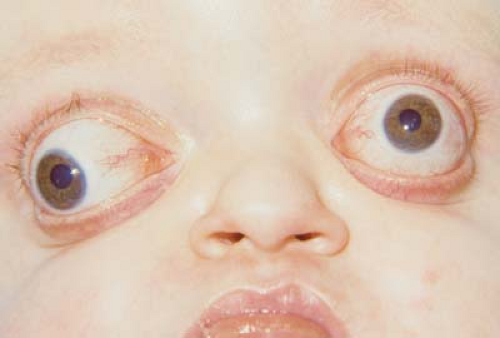 Figure 14.9 Apert Syndrome—Strabismus This child with the multiple craniosynostosis syndrome, Apert syndrome, shows significant exorbitism (Fig. 14.6) and strabismus. Syndactyly of the fingers and toes is a characteristic of Apert syndrome. The child has a large-angle right exotropia and hypotropia. This picture was taken in upgaze. Note the deficient elevation in both eyes, more so on the right. This may be due to anomalous or absent extraocular muscles. The patient is at risk for developing exposure keratopathy and amblyopia. Patching therapy is difficult due to the risk of corneal abrasion from the patch. This patient most likely will need advancement of the orbital bones. Following this surgery, strabismus surgery can be considered. |
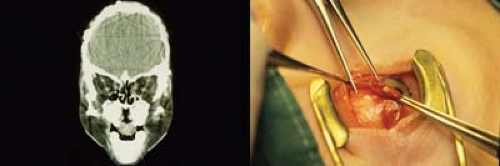 Figure 14.10 Apert Syndrome—Anomalous Muscles
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|