Corneal Dysgeneses and Degenerations
Jason S. Ehrlich MD, PhD
Glenn C. Cockerham MD
Kenneth R. Kenyon MD
Corneal dysgeneses and degenerations account for a broad spectrum of ocular abnormalities that range from clinical curiosities to sight-threatening anomalies. Historically, knowledge of these entities accrued through clinical study and examination of histopathologic specimens, but in the last two decades, significant progress has been made in understanding the genetics, molecular biology, and cellular biology of several of these disorders. This molecular research has increased our understanding not only of the individual diseases, but also the physiology and development of the cornea in its normal state.
Dysgeneses of the cornea are developmental disorders, sometimes inherited, resulting in congenital malformations. Corneal dysgeneses may be unilateral or bilateral and are nonprogressive. The central, peripheral, or entire cornea, as well as other ocular structures, may be affected and, not infrequently, associated systemic abnormalities are present. Corneal degenerations, in contrast, are historically described as having no developmental or hereditary pattern and may be unilateral or bilateral. They are frequently a manifestation of aging, inflammation, or environmental insult and, therefore, are not typically congenital. Degenerations most often begin in the peripheral cornea, although central vision may be affected. Inflammation is sometimes involved early in the degenerative process and may be accompanied by corneal vascularization. In some cases, such inflammatory processes may be associated with systemic disease (e.g., connective tissue disorders). Table 1 lists the corneal dysgeneses, and Table 2 lists the corneal degenerations.
TABLE 1. Corneal Dysgeneses. | |
|---|---|
|
TABLE 2. Corneal Degenerations | |
|---|---|
|
Corneal Dysgeneses: Abnormalities of Size and Curvature
Absence of Cornea
Complete absence of the cornea is rare, documented in sporadic case reports. It is associated with variable absence of other anterior ocular structures derived from surface ectoderm, and the eye consists of a sclera-like enclosure lined with neural ectoderm.1 The disorder is typically described in the literature as “congenital primary aphakia,” not a primary corneal dysgenesis. In 2006, a mutation in the FOXE3 fork head box transcription factor was shown to be responsible for cases of CPA in one family.2 Ultrasonography should aid in differentiating this entity from cryptophthalmos.
Microcornea
A normal adult corneal horizontal diameter (white to white) measures 11 to 12 mm horizontally and 10 to 11 mm vertically. Using the OrbScan II analyzer, Rufer et al. measured 390 patients (all Caucasian, ages 10 to 80) and found an average horizontal diameter of 11.71 +/- 0.42 mm.3 Microcornea is generally accepted as describing an adult cornea of less than 10 mm horizontal diameter or a newborn cornea of less than 9 mm. The microcornea is flatter than average, and patients are typically hyperopic, although cases of microcornea associated with myopia are described.4,5,6 Microcornea is often found along with other anterior segment developmental anomalies, and thus is described in a large number of inherited and sporadic conditions. Congenital cataract is an associated finding, and more rarely aniridia and anterior segment dysgenesis.7,8,9,10 When microcornea occurs along with anterior segment dysgenesis, angle-closure glaucoma may occur due to crowding or congenital abnormalities of the angle structures.
Importantly, microcornea may be a finding associated with systemic conditions of a genetic (Ehlers-Danlos, Marfan syndromes) or environmentally induced basis (fetal alcohol syndrome).11,12
Unsurprisingly, microcornea is transmitted both as autosomal dominant and recessive traits.13,14,15,16 Mutations in many genes have been described, including structural genes such as fibrillin-1 and connexin-50, transcription factors such as PAX-6 and MAF, and even in lens crystallins.17,18,19,20,21,22,23
The microcornea, although small, typically is clear and has normal corneal histology.
Megalocornea
Megalocornea typically refers to a horizontal corneal diameter of greater than 13.0 mm, although in some literature a value of 12.0 mm is employed. Patients with megalocornea are typically myopic and astigmatic (usually with-the-rule astigmatism). As in microcornea, the megalocornea is clear and with normal histology and thickness. Megalocornea can occur as a sole finding (simple megalocornea) or simultaneously with other anterior segment abnormalities, such as cataract or anterior megalophthalmos (see the subsequent text).24,25,26,27,28,29
Inheritance of megalocornea is often X-linked recessive, mapping to Xq21-q22, but no gene has yet been cloned for this locus.30,31,32 Affected males demonstrate nonprogressive, bilateral, symmetrically enlarged corneas, and carrier females can display subtly enlarged corneas.33 Autosomal dominant and numerous autosomal recessive syndromes are also described, including nearly 40 case reports of megalocornea-mental retardation (MMR) syndrome in which isolated megalocornea is the main ophthalmologic finding.34,35,36,37 In most of these genetic syndromes, megalocornea occurs as part of a syndrome of anterior megalophthalmos.
It is crucial for the ophthalmologist to distinguish benign megalocornea from the pathologically enlarged corneas of congenital glaucoma.38 In megalocornea, the cornea is typically clear, and no tears in Descemet’s membrane are observed. The optic nerve and intraocular pressure are usually normal, although ocular hypertension has been described in autosomal dominant megalocornea.39 In simple megalocornea, specular microscopy demonstrates a normal endothelial cell count, whereas the swollen cornea of congenital glaucoma shows a reduced number and abnormal morphology of endothelial cells.40
Anterior Megalophthalmos
In this hereditary condition, the entire anterior segment is enlarged, including the cornea, ciliary body ring, and the lens-iris diaphragm.41 The anterior chamber is abnormally deep, and the patient is often quite myopic due to improper lens position along the anterior-posterior axis.26 The iris demonstrates iridodesis and poor dilation, and pigmentary glaucoma has been reported, which is presumably due to the abnormally loose zonules that allow chronic rubbing of the lens against the posterior iris.27
Eyes with this condition often develop early cataract, and intraocular lens implantation can be difficult due to the abnormal anterior segment architecture.42 Refinement of intraocular lenses and phacoemulsification techniques and instrumentation have allowed more successful management of these patients.4,5,6,7,8,9,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46 Photorefractive keratectomy has also been successfully performed.47
Cornea Plana
In cornea plana, the corneal curvature is flatter than normal and often reaches levels as low as 20 to 30 D with a radius of curvature similar to the sclera.50,51 Peripheral scleralization of the cornea is almost always present, and limbal landmarks are typically obscured; therefore, the condition may simulate microcornea. A careful slit-lamp examination, however, will readily reveal the difference in corneal curvature between the two. In cornea plana, the anterior chamber is shallow by virtue of the low-corneal dome. Refractive abnormalities may vary depending on the globe dimensions and exact state of the corneal curvature.52 This condition may also feature concurrent anterior segment abnormalities, including iris colobomas, congenital cataract, and occasional posterior segment colobomas. The distortion of the cornea along with concomitant sclerocornea may lead to a decrease in corneal transparency.
Congenital cornea plana exists as both autosomal dominant and recessive forms.53,54 Genetic mapping studies in humans led to the discovery that the keratocan protein is responsible for the autosomal recessive form.55,56 Since 2001, numerous additional mutations have been described in different families.57,58,59,60 Keratocan is a leucine-rich proteoglycan, which is a significant component of the corneal stroma. An animal model of cornea plana has been created by knock-out of the mouse keratocan gene, and analysis suggests that keratocan mutations in humans result in larger, less organized collagen fibrils in the stroma, which leads to the disease phenotype.61,62
Mesenchymal Dysgeneses
Mesenchymal dysgenesis comprises a large spectrum of congenital disorders, which affect a range of tissues in the anterior segment. These historically have been known by a variety of names including mesodermal dysgenesis and anterior segment cleavage syndrome. A number of theories were advanced to describe this group of congenital abnormalities, which were all based on historical concepts of anterior segment embryogenesis. Recent developments in our knowledge of ocular embryology at the molecular level leads to the current hypothesis that mesenchymal dysgenesis largely reflects developmental arrest due to improper neural crest cell migration, signaling, and/or function, although anterior segment structures not derived from neural crest may also be affected.63,64,65,66,67,68 Neural crest cells migrate into the developing anterior segment in waves, and deviation from the normal pattern at any stage may lead to the clinically recognized syndrome. Although the last few years have seen very exciting discoveries in some of these syndromes (such as Axenfeld-Rieger, see the subsequent text), the molecular nature of others remains unknown.
Posterior Embryotoxon
Posterior embryotoxon is the simplest dysgenesis of the anterior segment periphery. The clinical finding is anterior displacement and enlargement of Schwalbe’s line. Schwalbe’s line is the peripheral terminus of Descemet’s membrane, which is not normally visible through the cornea. By slit-lamp microscopy, Schwalbe’s line in this case appears as an irregular, circumferential ridge on the posterior surface of the cornea just inside the limbus (Figure 1). The prevalence has been estimated from 6.8% to as high as 24.3% in a small Japanese study, but this has not been carefully studied.69,70 Posterior embryotoxon is associated with several serious genetic conditions in children, including most prominently Alagille syndrome; the phenotype in this disorder is characterized by serious congenital heart and liver disease and craniofacial anomalies.71,72 Some case series document posterior embryotoxon in as many as 90% to 95% of Alagille patients; thus, the ophthalmologist may be asked to assist in the diagnosis of this serious systemic condition. Posterior embryotoxon has also been reported as a significant finding in chromosome 22q11.2 deletion syndrome and Hermansky-Pudiak syndrome.73,74
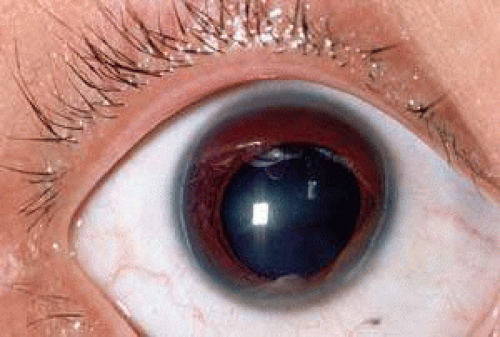 FIGURE 1. Posterior embryotoxon. Slit lamp photo demonstrates posterior embryotoxon, which is visible as a white line central to the limbus. |
Axenfeld-Rieger Type Syndromes
The terms Axenfeld anomaly, Axenfeld syndrome, Rieger anomaly, Rieger syndrome, Axenfeld-Rieger syndrome (and others: iris hypoplasia, iridogoniodysgenesis, familial glaucoma iridogoniodysplasia) all describe a spectrum of anterior segment mesenchymal dysgeneses involving the peripheral cornea, angle structures, and iris. The main ophthalmologic issue in many affected patients is the development of difficult-to-control glaucoma. We now understand that these disorders represent overlapping phenotypes and likely have the same molecular basis; the difference in phenotypic expression results from different point mutations in the identified loci and variable expressivity of the mutation in each patient’s unique genetic background. The consensus from those studying the field is that, in the future, clinicians and scientists should refer to the spectrum as Axenfeld-Rieger syndrome.65,75 For the sake of historical completeness, we describe the original phenotypes later.
Axenfeld Anomaly and Syndrome
Axenfeld anomaly was described as posterior embryotoxon accompanied by abnormal iris strands crossing the anterior chamber angle to attach to the prominent Schwalbe’s line.76 It has on occasion been reported in conjunction with Down’s syndrome and fetal alcohol syndrome.77,78 If glaucoma develops as a result of the angle obstruction and maldevelopment, the condition is called Axenfeld syndrome.79 This has been reported along with oculocutaneous albinism and, more rarely, with complex congenital anomalies.80,81,82 Goniotomy has long been the mainstay of surgical management of the congenital glaucoma.83
Rieger Anomaly and Syndrome
Rieger anomaly is present if hypoplasia of the anterior iris stroma is found with the changes typical of Axenfeld anomaly (Figure 2).84 Rieger anomaly is associated with glaucoma in approximately 60% of cases. Rieger syndrome is present when the eye anomaly is accompanied by skeletal abnormalities, such as maxillary hypoplasia, microdontia, and other limb and spine malformations.85,86,87,88 Marfan syndrome, Meckel’s diverticulum, and osteogenesis imperfecta have also been described in conjunction with Rieger syndrome.84,89,90,91
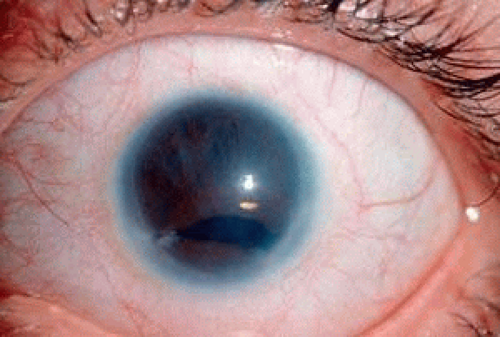 FIGURE 2. Rieger anomaly. Superior iris hypoplasia and corectopia is present in conjunction with posterior embryotoxon. |
Genetics and Molecular Biology of Axenfeld-Rieger Syndromes
In 1996 and 1997, two groups independently identified mutations in Pitx2, a homeobox transcription factor, as responsible for phenotypes in Axenfeld-Rieger syndrome.92,93 Subsequently, many novel mutations in Pitx2 were and continue to be identified in families with Axenfeld-Rieger class findings.94,95,96,97,98 Analysis using animal and tissue culture models indicates that specific mutations in different domains of the protein explain a significant amount of the phenotypic variation in the syndromes.99,100,101,102 In addition to the Pitx2 transcription factor, mutations in the Pax6 “master regulatory” gene have been identified,103,104 as have mutations in the FoxC1 transcription factor.105,106 A detailed review of the molecular biology is beyond the scope of this chapter, but the reader interested in the molecular biology and genetics of the Axenfeld-Rieger class of disorders is invited to examine several comprehensive reviews on the subject.107,108
Peters Anomaly (Congenital Central Corneal Opacity)
Peters anomaly is a congenital, central corneal opacification with absence of the posterior stroma, Descemet’s membrane, and endothelium in the region of the opacity. Two clinical variants have been recognized: Peters anomaly type I shows the typical nebular opacity in the pupillary axis, bordered by iris strands that cross the anterior chamber (Figures 3 and 4). The lens usually remains clear and is normally positioned. Associated anomalies, such as microcornea, sclerocornea, and infantile glaucoma may be present, but, for the most part, no other ocular or systemic abnormalities are present. In Peters anomaly type II, in contrast, the lens is abnormal either in position or transparency in addition to the central corneal opacity and iridocorneal synechiae. Centrally, the posterior cornea and lens may be adherent, and there may be an anterior polar cataract. This type is more frequently bilateral and almost every involved case shows severe ocular and systemic malformations. Associated abnormalities of the anterior segment include microcornea, microphthalmos, cornea plana, sclerocornea, colobomas, aniridia, and dysgenesis of the angle and iris.109,110 A “Peters-plus” syndrome has been described in which Peters anomaly occurs in conjunction with short status and brachydactyly.111,112,113
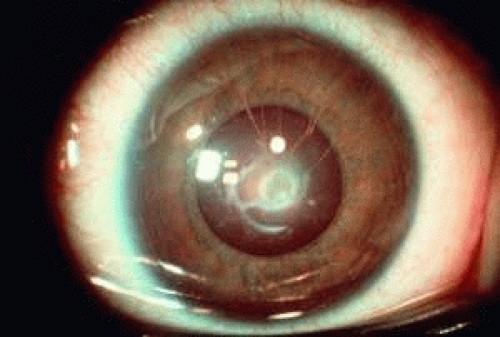 FIGURE 3. Peters anomaly. Iris strands bridge the anterior chamber to a central corneal opacity.
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|