6 Cornea and Sclera
CORNEAL HISTOLOGY
The cornea is the principal refractive element of the eye. Its anterior surface is covered by a layer of nonkeratinized stratified squamous epithelium five or six cells in thickness. The inconspicuous basement membrane of the epithelium rests on Bowman membrane, a feltwork of modified stroma. The bulk of the cornea is composed of paucicellular collagenous stroma that contains keratocytes and artifactual clefts. The posterior surface of the cornea is lined by the corneal endothelial cells. This delicate monolayer of cells rests on a thick Periodic acid-Schiff (PAS)-positive basement membrane called Descemet membrane, which it secretes (Fig. 1-8).
DEVELOPMENTAL ANOMALIES
The corneal epithelium is derived embryologically from surface ectoderm. The corneal endothelium and stroma are derived from successive waves of migrating neural crest cells.
Developmental anomalies of the cornea include microcornea, defined as <11 mm in greatest diameter, and megalocornea, which is >13 mm. Cornea plana is a bilateral familial trait (autosomal dominant or recessive) characterized by corneal flattening and peripheral opacification. The cornea in sclerocornea resembles sclera. The epithelium is thickened, Bowman membrane is absent, and the anterior third of the stroma is scarred and vascularized. Solid dermoids and complex choristomas are discussed in Chapter 5.
The Axenfeld-Reiger syndrome is a spectrum of developmental anomalies that affects the peripheral cornea, the iris, and the angle and has associated systemic findings. In the past, the terms angle cleavage syndrome and mesodermal dysgenesis were commonly applied to this disorder. Axenfeld-Reiger syndrome is important because about half of affected patients develop glaucoma. Prominence and anterior displacement of Schwalbe line into clear cornea, which is called posterior embryotoxon of Axenfeld, occurs in many patients (Fig. 6-1). Processes of iris stroma often bridge the angle and insert onto the posterior embryotoxon (Axenfeld anomaly). Patients may have iris stromal abnormalities including hypoplasia, and slit, multiple (polycoria), and false pupils (pseudocoria).
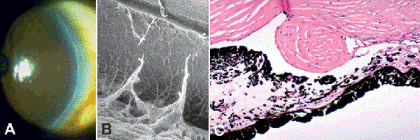
Fig. 6-1. Posterior embryotoxon. A. Prominent, anteriorly displaced line parallels limbus in clinical photo. B. Posterior embryotoxon is seen as ridge separating corneal endothelium and trabecular meshwork in scanning electron micrograph of infant eye. Ruptured iris processes bridge angle to embryotoxon. C. Large oval mound of connective tissue is interposed between the end of Descemet membrane (Schwalbe line) and the trabecular meshwork. This infant had multiple anterior segment anomalies. (B. SEM ×20, C. H&E ×50)
Axenfeld-Rieger syndrome is inherited as an autosomal dominant trait. Mutations in the FOXC1, PITX2, and RIEG2 genes have been found in the syndrome. Affected patients can have developmental defects of the face (maxillary hypoplasia) and teeth (anodontia, oligodontia, microdontia, and peg-like incisors) and redundant periumbilical skin. Shields has postulated that the ocular abnormalities in Axenfeld-Rieger syndrome are consistent with a developmental arrest, occurring late in gestation, of certain anterior ocular structures derived from neural crest.
Congenital corneal opacities occur in Peters anomaly, a developmental anomaly characterized by a concave defect in the central cornea that includes posterior stroma, Descemet membrane, and endothelium (Fig. 6-2). Histopathology shows a central concave defect in the posterior corneal stroma that often contains thickened abnormal lamellae. The endothelium and Descemet membrane are absent within the posterior ulcer, and there is a corresponding area of stromal edema and opacification. Bowman layer may be thickened or absent. The crystalline lens adheres to or is incarcerated within the posterior ulcer in some cases of Peters anomaly, and (Fig. 6-2C,D) iridocorneal adhesions often insert into the periphery of the posterior corneal defect. Peters anomaly can be caused by mutations in PAX6, PITX2, CYP1B1, or FOXC1 genes. Peters anomaly also occurs in fetal alcohol syndrome. This spectrum of developmental anomalies includes the posterior ulcer of von Hippel and posterior keratoconus. The posterior concavity is lined by Descemet membrane and endothelium in posterior keratoconus.
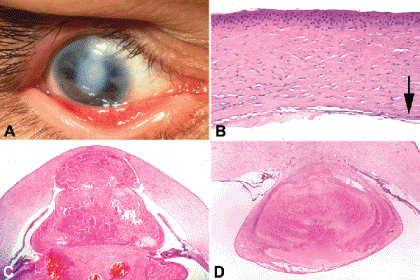
Fig. 6-2. Peters anomaly. A. Bands of iris stroma insert into the margin of a central corneal opacity forming iridocorneal adhesions. The lens was adherent to the posterior cornea centrally. Both eyes were affected. (Photo courtesy of Dr. Irving Raber, Wills Eye Institute.) B. The endothelium and Descemet membrane are absent in the region of a central “posterior ulcer.” Arrow denotes Descemet membrane at margin. The collagenous lamellae of the posterior stroma are thickened and irregular. Bowman membrane is absent and the epithelium is mildly thickened. A central corneal opacity was present clinically. C. Keratolenticular adhesion. Lens adheres to large defect in center of cornea. D. Peters anomaly with keratolenticular adhesion in child with fetal alcohol syndrome. The cataractous crystalline lens adheres to a posterior ulcer in the central cornea. The iris also attaches to the margin of the ulcer. (Case courtesy of Dr. Nongnart Chan, Philadelphia, Pennsylvania.) (B. H&E ×50, C. H&E ×10, D. H&E ×25)
CORNEAL INFLAMMATION
Acute Keratitis and Corneal Ulceration
The anterior surface of the cornea is an interface between the eye and the external environment. Mechanisms that protect the cornea from infection include the corneal epithelium, mucous strands secreted by conjunctival goblet cells that can ensnare microorganisms, and tears that can wash them away. In addition, the tears contain natural antimicrobial substances such as lactoferrin, lysozyme, and antibodies. The latter are made by lymphocytes and plasma cells that are always found in the stroma of the conjunctiva and the parenchyma of the lacrimal gland (Fig. 6-3).
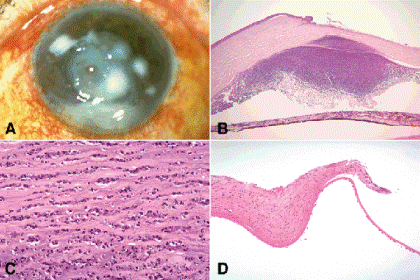
Fig. 6-3. Acute keratitis. A. Clinical photo of acute keratitis with ulceration and hypopyon. B. Anterior chamber deep to corneal ulcer contains hypopyon. Iris is flattened by neovascular membrane. C. Acute keratitis. Polymorphonuclear leukocytes and inflammatory debris fill clefts between the stromal lamellae. Many polys have pyknotic nuclei and early stromal necrosis is present. D. Descemetocele, acute keratitis. An intact layer of Descemet membrane persists in the bed of deep corneal ulcer. The anterior layers of the cornea have been destroyed by inflammation. (B. H&E ×25, C. H&E ×100, D. H&E ×50)
Normally, these mechanisms are remarkably effective. Corneal infection usually develops when these protective mechanisms are compromised, for example, when a contaminated foreign body perforates the epithelium and inoculates the corneal stroma, or when the epithelium is abraded or damaged by chronic bullous keratopathy. Finally, a few organisms, most notably Neisseria gonorrhoeae, have the ability to invade through an intact epithelium.
Infiltration of the stroma by polymorphonuclear leukocytes occurs in acute bacterial keratitis (Fig. 6-3C). The polys collect in the clefts between adjacent stromal lamellae. Digestive enzymes released by the dying inflammatory cells cause stromal necrosis, which has a smudged basophilic appearance in routine hematoxylin and eosin (H&E) sections. Collagenase made by the corneal epithelium may contribute to dissolution of the stroma. Organisms such as Pseudomonas also produce potent enzymes that contribute to corneal destruction. Pseudomonas keratitis is characterized by marked stromal edema and dissolution and rapid corneal perforation. The infection can also spread posteriorly into the sclera as a sclerokeratitis (Fig. 6-4).
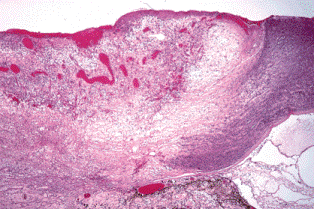
Fig. 6-4. Pseudomonas sclerokeratitis. Pseudomonas keratitis often extends posteriorly as an infectious scleritis. The acutely inflamed cornea (at right) appears blue, reflecting necrosis and heavy infiltration by polys. Proteolytic enzymes released by the Gram-negative rods have dissolved the limbal sclera. The angle is closed. (H&E ×20)
The epithelium, Bowman membrane, and the anterior stroma are lost in corneal ulcer. Corneal perforation results when the infection destroys the full-thickness stroma. Occasionally, a bulging dome of intact, elastic Descemet membrane called a descemetocele persists in the floor of the incipient perforation (Fig. 6-3D). Polymorphonuclear leukocytes collect in the lower part of the anterior chamber as a hypopyon, and also typically adhere to the posterior cornea directly beneath the ulcer. The endothelium is often damaged. The tissue Gram stain may disclose myriad microorganisms in the infected cornea or none at all. Bacteria are often found in relatively noninflamed parts of the stroma bordering the inflammatory infiltrate. Perforation or impending perforation of the cornea is an indication for emergent penetrating keratoplasty (corneal transplantation). Cyanoacrylate tissue adhesive often is used to seal corneal perforations. Although the “crazy glue” is lost during processing, its presence is marked by a tell-tale scalloped pattern on the anterior corneal surface.
Infectious pseudocrystalline keratopathy (Fig. 6-5) usually is caused by avirulent strains of streptococci, which proliferate in the relatively noninflamed stroma forming large interlamellar bacterial colonies that have a vaguely crystalline configuration on biomicroscopy. This relatively rare disorder typically occurs in corneal grafts after chronic therapy with corticosteroids. The bacterial colonies are sequestered by a glycocalyx whose formation is stimulated by the steroids.
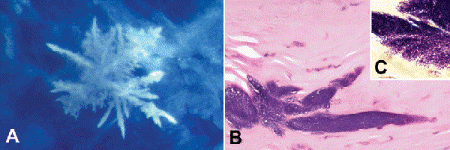
Fig. 6-5. Infectious pseudocrystalline keratopathy. A. Macrophoto shows radiating crystalline appearance of bacterial colony in cornea. B. Large basophilic colonies of relatively avirulent streptococci distend interlammelar clefts in relatively noninflamed part of the corneal stroma. C. Bacteria are Gram-positive. (B. H&E ×100, C. Tissue Gram stain ×250)
Fungal keratitis (Fig. 6-6) is rarer than bacterial keratitis, is more prevalent in the South, and often complicates corneal injuries by vegetable matter or steroid therapy in debilitated hosts. Eighty percent of corneal ulcers in the United States are caused by Aspergillus, Candida, or Fusarium species. Clinically, fungal ulcers often have a deep crater with raised edges, and may have smaller satellite lesions. The fungal hyphae readily permeate the stroma and can perforate Descemet membrane and invade the anterior chamber, placing the patient at risk for fungal endophthalmitis (Fig. 6-6D,E).
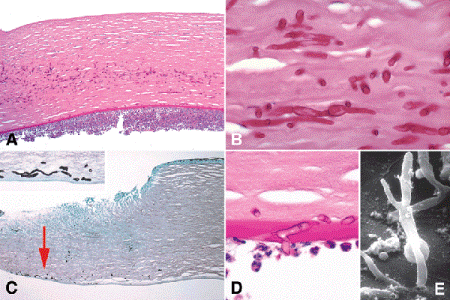
Fig. 6-6. Fungal keratitis. A. Periodic acid-Schiff stain discloses numerous hyphae in mid stroma, seen at higher magnification in part B. The corneal epithelium is absent. A hypopyon adheres to the posterior cornea. C. Deep hyphae, fungal keratitis. The corneal epithelium and the anterior stroma are absent in the ulcerated area at left. Arrow points to GMS-stained hyphae in the deep stroma near Descemet membrane. Hyphae are seen at higher magnification in inset. A superficial scraping of the ulcer bed was negative for fungus. D. PAS-positive septate fungal hypha perforates Descemet membrane and invades anterior chamber, which contains polymorphonuclear leukocytes. E. Scanning electron micrographs shows branching fungal hyphae that have invaded anterior chamber. (A. PAS ×25, B. PAS ×250, C. GMS ×50; Inset, GMS ×100, D. PAS ×250, E. SEM ×640)
Fungi may not be detected in superficial smears or scrapings because the hyphae are often found deep in the bed of the ulcer, or in the relatively viable stroma of the ulcer wall (Fig. 6-6C). Hence, corneal biopsy may be required to establish the diagnosis. Fungi and yeast are best shown by the PAS stain or special stains for fungus such as the Gomori methenamine silver (GMS) impregnation stain. The nonspecific fluorochrome calcofluor white binds to cellulose and chitin in cell walls of fungi, pneumocystis, and acanthamoebae. It can be used to detect fungi, but requires an ultraviolet microscope.
M. tuberculosis, M leprae, and several strains of atypical mycobacteria such as M. chelonae rarely cause corneal infection. Special stains for acid-fast organisms and microbiological cultures are used to diagnose mycobacterial keratitis. The diagnosis is often unsuspected and is frequently delayed. Organisms are often quite numerous in atypical mycobacterial infection.
VIRAL KERATITIS
Herpes simplex (HSV) keratitis (Fig. 6-7) is the most common corneal infection that causes visual loss in the United States and Europe. Most cases of herpetic keratitis are caused by HSV Type I, which causes infection above the waist. The classic clinical manifestation of early ocular herpesvirus infection is dendritic keratitis, a superficial ulcerating infection of the corneal epithelium that has a characteristic branching configuration (Fig. 6-7A). Viral cultures are positive in 75% of cases, and smears show Cowdry type A intranuclear viral inclusions in the infected epithelial cells. The primary epithelial infection tends to be self-limited, lasting 7 to 10 days with complete recovery and disappearance of virus from the primary site of infection. However, recurrences are quite common because the virus remains in a latent state in the trigeminal ganglion where its DNA has been incorporated into the genome of the neurons. Poorly understood trigger mechanisms reactivate the virus, which travels down the sensory nerve to cause overt recurrent disease. One in four cases of primary HSV keratitis recur. The recurrence rate rises to 50% after an initial recurrence.
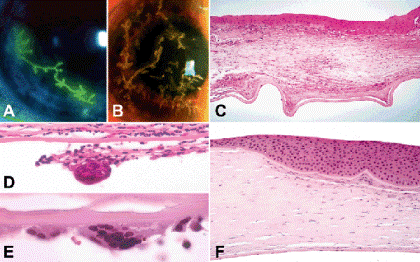
Fig. 6-7. Herpes simplex keratitis. A. HSV dendritic keratitis. Branching Herpes simplex viral dendrite is full-thickness ulceration with terminal bulbs. Dendrite is stained with fluorescein dye. B. VZV dendritic keratitis. Dendritiform lesions of varicella-zoster keratitis are composed of heaped-up epithelium. (Both photos courtesy of Dr. Peter Laibson, Wills Eye Institute.) C. Chronic herpetic stromal keratitis. The stroma is thin, scarred and chronically inflamed. The inflammatory infiltrate contains lymphocytes, plasma cells and epithelioid histiocytes. Bowman membrane is largely destroyed and the epithelium is irregularly thickened. Descemet membrane is folded. D, E. Giant cell reaction to Descemet membrane, Chronic HSV keratitis. Although characteristic, this finding is not pathognomonic for HSV. F. Compensatory epithelial hyperplasia, chronic HSV keratitis. The epithelium is markedly thickened overlying an area of stroma loss. Bowman membrane persists in the area of stromal thinning. (C. H&E ×50, D. H&E ×100, E. H&E ×250, F. H&E ×50)
A more serious, potentially blinding form of ocular herpesvirus infection affects the corneal stroma. Although the pathogenesis of herpes stromal keratitis is not entirely understood, an immunological response to viral antigens shed from infected corneal epithelial or endothelial cells probably is basis for nonulcerative HSV disciform keratitis. The necrotizing form of HSV stromal keratitis actually may involve viral invasion and low-grade replication in the stroma.
Light microscopy of corneal buttons with chronic herpes stromal keratitis typically reveals scarring and vascularization of the stroma and infiltration by chronic inflammatory cells (Fig. 6-7C). The presence of lymphocytes, plasma cells, and histiocytes in the inflammatory infiltrate in many cases provides evidence for an immunological response. The inflammatory infiltrate frequently includes epithelioid histiocytes. These can occur anywhere in the stroma, but they characteristically coalesce to form giant cells near Descemet membrane (Fig. 6-7D,E). A giant cell reaction to Descemet membrane is not pathognomonic for chronic HSV keratitis, but it is highly suggestive. In many cases, there is thinning and scarring of the chronically inflamed cornea with loss of Bowman membrane and anterior stroma. Progressive stromal loss can lead to corneal perforation in chronic herpetic keratitis. Compensatory hyperplasia of the corneal epithelium is often found in areas of extensive stromal loss (Fig. 6-7F). Viral inclusions are not found light microscopically in the stroma in herpes stromal keratitis. Virions and viral antigens have been detected electron microscopically and immunocytochemically, however.
Varicella zoster virus (VZV), another member of the herpesvirus family, also causes a dendritic keratitis. Zoster dendrites lack the rounded terminal bulbs that typically occur at the end of the branches of HSV dendrites, and are not full-thickness epithelial ulcerations as HSV dendrites are (Fig. 6-7B).
INTERSTITIAL KERATITIS
In a generic sense, the term interstitial keratitis refers to any nonulcerative inflammation of the corneal stroma. (HSV disciform keratitis is a form of interstitial keratitis.) Other causes of stromal or interstitial keratitis include tuberculosis and leprosy, protozoan parasites such as Acanthamoeba, onchocercal microfilaria, systemic disorders such as sarcoidosis, Hodgkin disease and mycosis fungoides, foreign bodies such as caterpillar setae and plant material, and drugs including gold and arsenic.
In common parlance, however, the term interstitial keratitis generally connotes the severe stromal keratitis that affects patients with congenital syphilis during the first or second decade. Acute luetic interstitial keratitis is marked clinically by a “salmon patch” of intense stromal vascularization and severe photophobia. Lymphocytes infiltrate the edematous stroma. The late sequelae of old syphilitic interstitial keratitis can be relatively subtle: they include a faint diffuse nebulous opacification of the stroma and residual nonperfused ghost vessels located deep in the corneal stroma (Fig. 6-8A). The ghost vessels are best disclosed by the biomicroscopic technique of sclerotic scatter.

Fig. 6-8. Chronic luetic interstitial keratitis. A. Clinical photo shows faint nebulous opacification of the cornea. B. Double arrow denotes vessels deep within noninflamed corneal stroma. Single arrow points to guttate excresence on irregularly thickened Descemet membrane. C. Arrow in clinical photo denotes relucent mass of hypertrophic Descemet membrane material on the posterior cornea. D. Photomicrograph shows thick cylinder of PAS-positive basement membrane material studded with guttate excrescences on irregularly thickened Descemet membrane. (B. PAS ×100, C. Clinical photo courtesy of Dr. Irving Raber, Wills Eye Institute, D. PAS ×100)
Histopathologically, Bowman membrane usually is absent. The stroma is free of inflammation, and fine vessels that usually do not contain blood are seen in the posterior third of the stroma, often just anterior to Descemet membrane (Fig. 6-8B). Part of the stroma may have a rarefied vacuolated appearance and will stain intensely for acid mucopolysaccharide. Descemet membrane is often thickened and may be studded with irregular guttate excrescences (Fig. 6-8B). The thickening of Descemet membrane may be massive. Relucent strands and networks of basement membrane material are found on the posterior corneal surface in exceptional cases (Fig. 6-8C,D).
Luetic interstitial keratitis occasionally develops in acquired syphilis, in which corneal involvement is typically unilateral and sectoral. The association of nonluetic interstitial keratitis and vestibuloauditory symptoms is called Cogan syndrome. Cogan syndrome is thought to be an autoimmune disorder.
PARASITIC KERATITIS
Protozoan parasites including microsporidians and the freshwater amebas Acanthamoeba castellanii and A. polyphaga cause unusual corneal infections. Originally reported in patients with HIV/AIDS, microsporidial keratoconjunctivitis has occurred in immunocompetent individuals as well. The parasite can infect the epithelium of the cornea and conjunctiva or the corneal stroma (Fig. 5-11B).
Acanthamoeba keratitis was initially reported in 1974, and is closely linked to the development and widespread use of soft contact lenses. Acanthamoeba keratitis classically afflicts soft contact lens wearers who use contaminated homemade saline solutions or wear their lenses while swimming or bathing in hot tubs. The keratitis is usually severely painful. Although many cases respond to medical therapy, penetrating keratoplasty (corneal transplantation) may be necessary. The initial stages of acanthamoeba keratitis can be confused with herpetic keratitis if clinical suspicion is low. A ring or annular infiltrate is a characteristic feature of acanthamoeba keratitis, but usually develops in the later stages of the disease (Fig. 6-9A).
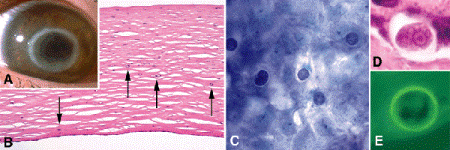
Fig. 6-9. Acanthamoeba keratitis. A. Clinical photo shows characteristic late ring infiltrate. B. Arrows denote amebic cysts in minimally inflamed corneal stroma. The epithelium is absent. Acanthamoeba cysts are seen in epithelial scraping stained with Giemsa (C) and sections stained with H&E (D) and calcofluor white (E). Cytoplasmic retraction from cyst wall and round nucleus with distinct nucleus are evident in part D. (A. Clinical photo courtesy of Dr. Elizabeth Cohen, Wills Eye Institute, B. H&E ×50, C. Giemsa ×100, D. H&E ×250, E. Calcofluor white ×250)
Microscopically, acanthamoeba keratitis is often marked by focal detachment or desquamation of the corneal epithelium. In contrast to bacterial or fungal keratitis, the anterior stroma in many cases contains only a relatively sparse infiltrate of inflammatory cells, mainly polys and macrophages (Fig. 6-9B). Stromal necrosis is usually inconspicuous, and stromal vascularization generally is not observed despite a lengthy course that may last months prior to keratoplasty. Amebic cysts in the stroma and epithelium are readily found in sections or smears stained with Giemsa or H&E (Fig. 6-9B–D). The chitinous walls of the acanthamoeba cysts are round or oval, and stain with PAS, GMS, and Giemsa. The cytoplasm typically retracts from the cyst wall in tissue sections. The cysts contain a round nucleus with a distinct nucleolus (Fig. 6-9D). Trophozoites are usually difficult to identify because they resemble macrophages. Intense stromal keratitis with localized abscess formation and palisading granulomatous inflammation can be observed in the late stages of the disease.
Acanthamoeba keratitis can be diagnosed using corneal biopsies, smears, or confocal microscopy. In smears, the organisms are most readily found within sheets of epithelial cells. The nonspecific fluorochrome calcofluor white can disclose cysts during the rapid screening of smears but requires an ultraviolet fluorescent microscope (Fig. 6-9E). Acanthamoebas are cultured on agar plates overlain with a layer of E. coli. The amebas produce diagnostic linear tracks on the surface of the plates as they graze on the bacteria.
Onchocerciasis is a major cause of blindness in parts of Africa and South and Central America. This infestation by the parasite Onchocerca volvulus is called river blindness because its vector, the black simulian fly, breeds in swift-running mountain streams. Pairs of adult Onchocerca breed in characteristic nodules in the skin of infested individuals. The female worm releases myriad microfilariae that migrate to all parts of the body. Necrotic microfilariae incite focal inflammation. Microfilariae are often found in the corneal stroma where they produce keratitis with a characteristic nummular (coin-shaped) pattern. Secondary closed-angle glaucoma caused by anterior uveitis is a major cause of blindness in endemic areas. Chorioretinal degeneration, also an allergic reaction to the parasite, is another important cause of blindness. Slit lamp examination may disclose microfilariae in the anterior chamber in heavily infested patients. The tiny worms are photophobic, and they quickly swim behind the iris when the lamp is turned on. Patients are treated by the surgical removal of parasitic nodules and with the antihelminthic drug ivermectin.
PERIPHERAL CORNEAL ULCERS
Although bacterial and fungal ulcers usually involve the central cornea, a variety of ulcerations affect its periphery. Most important are the peripheral corneal ulcerations that may herald the presence of a systemic vasculitis such as Wegener granulomatosis, periarteritis nodosa, systemic lupus erythematosus, or rheumatoid arthritis.
A rapidly progressive, painful, ulcerative keratitis, Mooren ulcer begins peripherally and extends across the cornea. The margin of the ulcer has a characteristic overhanging configuration (Fig. 6-10). Mooren ulcer usually is a unilateral disease of elderly patients in the United States. A severe, bilateral form of the disease affects young individuals in Africa. Autoimmunity is suspected in the pathogenesis of Mooren ulcer. Some cases have been linked to chronic hepatitis C infection.
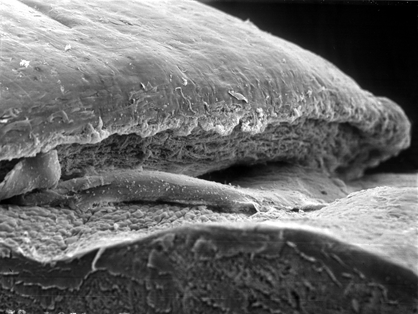
Fig. 6-10. Mooren ulcer. Scanning electron micrograph shows characteristic overhanging margin of ulcer. (SEM ×125)
Terrien ulcer is a slowly progressive bilateral trough-like thinning of the stroma that begins superiorly in men. The epithelium remains intact but Bowman membrane and the superficial stroma are lost. Vascularization and occasional lymphocytes and plasma cells are found in the ectatic stroma.
Toxins produced by staphylococci cause infiltrates and superficial ulcerations in the peripheral cornea.
Pterygium (Fig. 6-11) is a degenerative disease of the cornea characterized by a wedge-shaped ingrowth of conjunctival tissue that slowly invades the peripheral cornea. The nasal limbus is usually affected, and both eyes are often involved. The name pterygium reflects the fanciful resemblance of the vascularized ingrowth to the membranous wing of an insect (pter is the Greek word for wing). Pterygium initially is a cosmetic blemish, but it can cause visual loss if it invades the pupillary axis. Pterygia arise from limbal conjunctiva exposed to light in the interpalpebral fissure. Prolonged exposure to UV-B is thought to play an important pathogenetic role by causing DNA mutations with resultant loss of cell cycle regulation. Increased expression and mutations in tumor-suppressing transcription factor p53 occur in pterygium cells but do not appear to be a prerequisite for pterygium formation. Focal limbal stem cell deficiency probably is an important factor in the transdifferentiation of cornea epithelium into conjunctival-like tissue. Pterygia are more common in southern latitudes and typically occur in individuals who work outdoors. Other environmental factors such as wind or dust could play a pathogenic role.
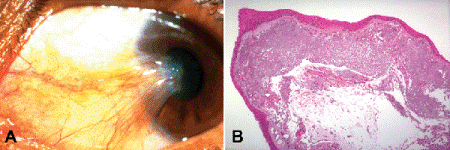
Fig. 6-11. Pterygium. A. Wedge-shaped ingrowth of conjunctival tissue invades the periphery of the nasal cornea. B. There is extensive actinic elastosis of the substantia propria in this example. (A. Clinical photo courtesy of Dr. Peter Laibson, Wills Eye Institute, B. H&E ×50)
Histopathologically, pterygia resemble an ingrowth of conjunctiva onto the periphery of the cornea. Bowman membrane is absent, and the stroma shows increased vascularity. Actinic elastosis of collagen is often present but may be relatively inconspicuous or absent in some cases. The epithelium usually resembles conjunctival epithelium. Epithelial hyperplasia and even dysplasia occur rarely. Pterygia should be examined histopathologically to exclude the possibility of an unsuspected neoplasm. In addition to squamous malignancies, amelanotic melanomas occasionally are misdiagnosed clinically as pterygia.
OTHER CORNEAL DEGENERATIONS
Calcific band keratopathy is a superficial opacity that extends across the part of the cornea exposed in the interpalpebral fissure (Fig. 6-12). It is caused by calcification of Bowman membrane and the anterior stroma. A clear interval separates the band of superficial calcification from the limbus, and the opacity usually contains small circular holes. Calcific band keratopathy can complicate chronic uveitis or long-standing glaucoma, and also complicates systemic disorders of calcium metabolism including hypercalcemia, vitamin D intoxication, milk-alkali syndrome, hypophosphatemia and Fanconi syndrome. Histopathology discloses basophilic granules in Bowman membrane and the superficial stroma. The alizarin red and von Kossa stains for calcium are positive. The chelating agent EDTA is often used to treat calcific band keratopathy. A noncalcific form of band keratopathy related to chronic actinic keratopathy also occurs. The granules are larger and stain positively with the Verhoeff-van Gieson elastic stain (Fig. 6-12C).
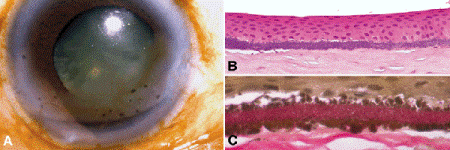
Fig. 6-12. Band keratopathy. A. Clinical photo shows band of superficial opacification involving the interpalpebral part of the cornea. B. Fine basophilic granules of calcium stipple Bowman layer. C. Elastic stain highlights larger granules of actinic elastosis in case with noncalcific component. (B. H&E ×100, C. Verhoeff-Van Gieson elastic stain ×250)
Chronic actinic keratopathy (Fig. 6-13) and climatic droplet keratopathy are two of many names applied to a degenerative disorder that is marked by the deposition of multiple spherules of yellow hyaline material in the anterior cornea. The disorder is also called spheroidal degeneration and Labrador keratopathy. The degeneration is particularly prevalent in areas where intense glare is reflected from ice (Labrador) or sand (the Dahlac Islands in the Red Sea). Other synonyms such as oleoguttate dystrophy and degeneratio sphaerularis elaoides stress the resemblance of the corneal deposits to droplets of olive oil. In H&E-stained sections, the drop-like deposits of amorphous hyaline material usually are light gray or amphophilic. The material stains intensely with the Verhoeff-van Gieson elastic stain, and the positive reaction is not abolished by pretreatment with elastase such as the elastotic degeneration in pinguecula and pterygium. The hyaline deposits autofluoresce under ultraviolet light and are said to behave similarly when illuminated with the slit lamp’s cobalt blue filter. In some cases, tiny globules of hyaline material are concentrated in Bowman membrane, mimicking calcific band keratopathy. Concurrent corneal scarring can severely decrease visual acuity in advanced cases.
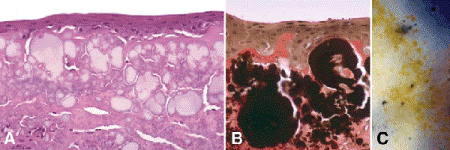
Fig. 6-13. Chronic actinic keratopathy. A. Anterior stroma contains amphophilic deposits of amorphous hyaline material. B. Material stains shows intensely with elastic stain. C. Material in scarred cornea resembles droplets of olive oil. (A. H&E ×100, B. Verhoeff-Van Gieson elastic stain ×125)
Salzmann nodular degeneration was once called Salzmann nodular dystrophy. It is now recognized that this typically unilateral disorder is not heritable and is best classified as a secondary degenerative process of uncertain cause. Clinically, the corneal epithelium is focally elevated by white mounds of dense collagenous connective tissue. Salzmann nodular degeneration resembles a massive focal pannus histopathologically. Mounds of relatively acellular hyaline connective tissue elevate the corneal epithelium anterior to the plane of Bowman membrane, which may be destroyed.
Lipid deposition occurs in the peripheral corneal stroma in arcus senilis (Fig. 6-29A) or gerontoxon, a relatively common aging change. Fat stains performed on frozen sections disclose a concurrent deposit in the perilimbal sclera which is inapparent clinically. Similar corneal deposition in a man younger than age forty may signify a hyperlipemic state that can predispose to cardiovascular disease. Lipid keratopathy is a secondary phenomenon caused by lipid deposition in a heavily vascularized stroma.
A corneal keloid (Fig. 6-14) is a hypertrophic scar that massively thickens the corneal stroma. Elevated and exposed, the corneal epithelium on the surface of the keloid typically undergoes epidermalization and transforms into opaque skin-like tissue. Large corneal keloids often develop in patients who have corneal staphylomas. In such cases, the posterior surface of the cornea is lined by atrophic remnants of iris, mainly iris pigment. A staphyloma is an ectasia (area of thinning) lined by uveal tissue. Uveal is derived from the Latin word uva meaning grape. Staphyle means a “bunch of grapes” in Greek. The latter prefix signifies the involvement of uveal tissue. Corneal staphylomas and keloids are common sequelae of keratomalacia and measles keratitis in underdeveloped countries.
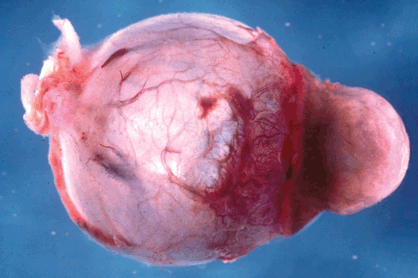
Fig. 6-14. Corneal staphyloma. The cornea is massively thickened by a hypertrophic scar (corneal keloid) that was lined internally by atrophic iris. The eye was enucleated from a child in Africa.
The tear film functions as the true anterior surface of the cornea, whose health depends on an adequate supply of properly constituted tears. Disorders of tear production and composition include deficiencies in the aqueous component, the mucinous wetting agent produced by the goblet cells, or abnormalities in the most superficial layer of lipid. Minor degrees of ocular drying are encountered often in clinical practice. If an eye is totally dry, the cornea becomes opaque.
Keratoconjunctivitis sicca is marked by corneal drying, superficial punctate keratopathy (punctate staining), and filamentary keratitis composed of strands of detached corneal epithelium and mucous. Keratoconjunctivitis sicca and xerostomia are characteristic features of the autoimmune disorder Sjøgren syndrome. Drying of the mouth and eyes is caused by infiltration and destruction of the acini of the salivary and the main and accessory lacrimal glands by T lymphocytes. Myoepithelial islands persist in the lymphoid infiltrate (lymphoepithelial lesion of Godwin). About 10% of affected patients develop malignant lymphoma.
Corneal epithelial keratinization and epidermalization occur in severe vitamin A deficiency. Patients have xerophthalmia and night blindness caused by deficient rod photopigment. A process of bland corneal melting called keratomalacia frequently leads to corneal perforation (Fig. 6-15). Malnourished children in underdeveloped countries and vitamin A–deficient alcoholics in the United States are at risk. Bitot spot, a clinical marker for xerophthalmia, is an elevated dry area of epithelium with a foamy appearance.
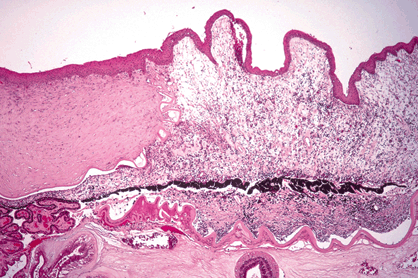
Fig. 6-15. Keratomalacia secondary to avitaminosis A. Incarcerated iris plugs sharply margined perforation in center of noninflamed cornea. Avitaminosis A was caused by dietary deficiency in chronic alcoholic. (H&E ×25)
A corneal delle (pl. dellen) is a focal area of corneal thinning with superficial surface ulceration, which is located central to an elevated lesion at the limbus. Dellen probably are caused by focal dehydration of the corneal stroma related to deficient focal corneal wetting.
Neurotrophic or neuroparalytic keratopathy is a corneal epitheliopathy that may complicate V nerve lesions or corneal hypesthesia caused by herpetic eye disease or herpes zoster ophthalmicus. The keratopathy develops rapidly after surgical sectioning of the trigeminal nerve interrupts poorly understood trophic factors. Bland corneal ulceration may develop.
The term pannus is applied to a flat superficial scar of the anterior cornea (Fig. 6-16). Two types of pannus are recognized histopathologically. The paradigmatic inflammatory pannus occurs in trachoma and is marked by a subepithelial ingrowth of inflamed fibrovascular tissue from the limbus, which destroys Bowman membrane. Degenerative pannus occurs in chronically edematous corneas with bullous keratopathy. Microscopically, a layer of connective tissue is found interposed between the base of the epithelium and Bowman membrane, which remains intact (Fig. 6-16B). In contrast to an inflammatory pannus, the subepithelial scar of degenerative pannus does not necessarily grow in from the limbus. Cells, which probably are stromal fibroblasts, migrate into the space between Bowman membrane and the detached epithelium and synthesize collagen. This fibrous scar forms the pannus and may alleviate the painful bullous keratopathy at the expense of visual acuity. In rare instances, amyloid is deposited secondarily beneath the epithelium.
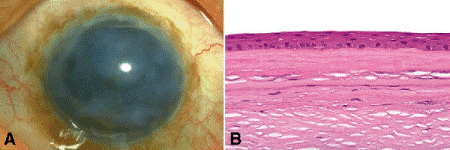
Fig. 6-16. Degenerative pannus, chronic corneal edema. A. Chronically edematous cornea is opacified by subepithelial fibrosis. B. A thick layer of relatively acellular connective tissue is interposed between the corneal epithelium and intact Bowman membrane. (B. H&E ×100)
Keratoconus is an enigmatic bilateral degenerative disorder characterized by progressive thinning or ectasia of the central stroma that imparts a conical configuration to the cornea (Fig. 6-17). Keratoconus usually presents around puberty with visual loss caused by severe astigmatism. Most cases of keratoconus do not appear to be inherited, but the degeneration can complicate several systemic disorders including trisomy 21, Ehlers-Danlos syndrome, Leber congenital amaurosis, and atopic dermatitis. Keratoconus occurs in approximately 5% of patients with Down syndrome and frequently is associated with the acute onset of severe corneal edema called corneal hydrops. Keratoconus in patients with atopic dermatitis, Down syndrome, and Leber congenital amaurosis, a congenital photoreceptor degeneration, could be related to forceful eye rubbing or the oculodigital reflex. The latter is a behavioral pattern seen in visually and mentally handicapped children who repeatedly strike their eyes with their thumbs in order to mechanically induce flashes of light or phosphenes.
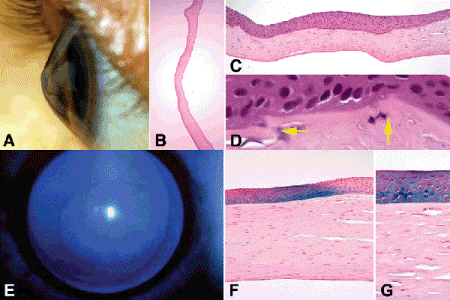
Fig. 6-17. Keratoconus. A. Clinical photo shows conical shape of cornea. B. Sectioned ectatic cornea has wavy configuration. C. Photomicrograph shows severe thinning of apical stroma, compensatory hyperplasia of the epithelium and multiple dehiscences in Bowman membrane. D. Arrows point to characteristic dehiscences in Bowman membrane. E. Cobalt blue illumination highlights Fleischer ring surrounding apex of cone. F, G. Iron stain of Fleischer ring shows focal deposition of iron in corneal epithelium surrounding cone. (B. H&E ×5, C. H&E ×25, D. H&E ×250, F. Iron stain ×100, G. Iron stain ×250)
The consistency of the cornea in keratoconus is abnormal; the sectioned cornea almost invariably assumes an irregular wavy configuration on a microslide, which immediately suggests the diagnosis (Fig. 6-17B,C). Higher magnification discloses central or apical thinning of the stroma, which can be reduced to less than one-tenth normal thickness in exceptional cases. Characteristic dehiscences, which typically have a wavy configuration, occur in Bowman membrane. These probably are pathognomic for keratoconus and serve to confirm the diagnosis histopathologically (Fig. 6-17C,D). Some cases exhibit apical scarring with increased fibroblastic activity and new collagen production. The corneal epithelium usually is intact and irregular in caliber with areas of both thinning and compensatory hyperplasia. Descemet membrane usually is thin, and the endothelium is well preserved. A rupture in Descemet membrane is found if acute hydrops has occurred. If the hydrops had resolved prior to corneal transplantation, the gap in Descemet membrane is lined by a thin layer of new Descemet membrane synthesized by endothelial cells that have migrated into the defect. Special stains show iron deposition in the corneal epithelium encircling the cone. This deposit is evident clinically as a Fleischer ring, one of the several eponymic iron lines of the cornea (Fig. 6-17E–G). Other iron lines unassociated with keratoconus include the Hudson-Stähli line (across the low third of the cornea), the Stocker line (at the advancing head of a pterygium), and the Ferry line (next to a filtering bleb). The basic defect in keratoconus is uncertain but may be related to abnormal degradation of the corneal extracellular matrix. Abnormal levels of tissue metalloproteinase inhibitors and alcohol dehydrogenase have been identified in some cases.
Pellucid degeneration of the cornea resembles keratoconus histopathologically but is located in the periphery of the cornea.
CORNEAL DYSTROPHIES
Introduction
In classic ophthalmic usage, the term dystrophy usually denotes an inherited, relatively symmetric bilateral disease that is unassociated with vascularization or inflammation in its early stages. The pathology is or appears to be localized to an ocular tissue. Dystrophies usually are not evident at birth, but become clinically evident later in life. The etymological derivation of the word dystrophy is outdated because it refers to poor nutrition. The term usually is applied to hereditary diseases of the cornea and macula.
Advances in molecular biology have markedly increased our understanding of these rare, interesting disorders. Several corneal dystrophies currently are known to be caused by allelic mutations in the gene encoding a single corneal protein. Macular corneal dystrophy (MCD) appears to be the ocular manifestation of an otherwise innocuous systemic enzyme deficiency. Meesman epithelial dystrophy is caused by an abnormality in the genes for proteins that are only expressed in the corneal epithelium.
Corneal dystrophies are classified topographically as superficial, stromal, and endothelial based on the location of the clinical and pathologic findings. A new classification system called the IC3D Classification of Corneal Dystrophies was published in 2009. The new classification incorporates many aspects of the traditional definitions of corneal dystrophies with new genetic, clinical, and pathologic information. This new classification can be upgraded in the future as our understanding of molecular genetics increases.
Superficial Corneal Dystrophies
Meesman dystrophy (Fig. 6-18A) is a relatively benign autosomal dominantly inherited disorder of the corneal epithelium. The eponym Stocker-Holt dystrophy is applied to a somewhat similar disorder. Clinically, Meesman dystrophy is characterized by the presence of myriad small punctate vacuoles in the corneal epithelium, which are best seen in retroillumination. Fluorescein dye typically pools in the vacuoles that have migrated to the corneal surface. Although recurrent epithelial erosions are possible, good vision is the rule. Histopathologically, both the epithelium and its basement membrane are thickened, and the epithelium has a disorderly appearance. In addition to small intraepithelial cystoid spaces, the epithelium contains cells that have a hyalinized appearance. Electron microscopy has disclosed intracellular aggregates of fibrillogranular “peculiar substance” composed of mutated cytokeratin. Meesman corneal dystrophy is caused by mutations in either of the genes (KRT3 or KRT12) that encode cornea-specific cytokeratins. The mutations severely impair cystoskeletal function and cause increased fragility of the corneal epithelium.
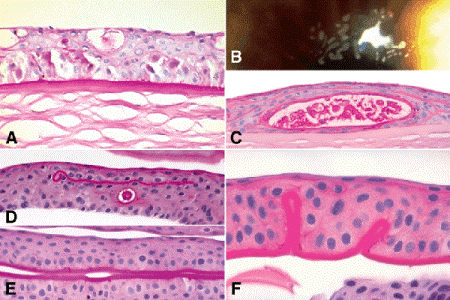
Fig. 6-18. Epithelial dystrophies. A. Meesman epithelial dystrophy. The epithelium is thickened and contains small cystoid spaces. The epithelial basement membrane is markedly thickened. B. Cogan microcystic dystrophy. Slit lamp discloses intraepithelial deposits of putty-like cellular debris. C. Photomicrograph of microcystic dystrophy shows devitalized cellular debris trapped by duplication of the epithelium. D–F. Map-dot-fingerprint dystrophy. Photomicrographs of corneal scrapings show small intraepithelial cyst and intraepithelial segment of basement membrane (D) and marked thickening of epithelial basement membrane (E). Folds of thickened basement membrane protrude into corneal epithelium. (A. PAS ×250, C. PAS ×100, D. PAS ×100, E. PAS ×100, F. PAS ×250)
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


