Conjunctival Diseases
Rudolph S. Wagner
CONJUNCTIVAL DISEASES
The conjunctiva is a durable mucous membrane composed of bulbar and palpebral components, which provides the primary barrier for protection of the eye. Making a definitive diagnosis for a pediatric patient presenting with signs and symptoms of disease in the conjunctiva can be challenging. This thin membrane is subject to neoplasms, vascular and structural abnormalities, environmental hazards, and is affected in many systemic diseases. Conjunctivitis in the pediatric patient can be mimicked by nasolacrimal duct obstruction and caused by allergies, bacteria, and viruses. Because antimicrobial cultures take time and are not always accurate, the diagnosis and treatment of conjunctivitis are often based on the physician’s knowledge regarding the current literature on likely pathogens and clinical experience.
Structure and Function
The conjunctiva originates from the superficial ectoderm of the early embryonic disc, which also gives rise to skin, corneal epithelium, cilia, glands, lacrimal glands, and the nasolacrimal system (1). It is a translucent vascular mucous membrane that is rich in immune components. Being firmly attached to the eyelid along the tarsus (palpebral) starting at the mucocutaneous junction and terminating 1 mm anterior of the corneal limbus (bulbar), the conjunctiva forms a cul-de-sac with its contours, creating the fornices on the superior, inferior, and lateral boundaries. Medially the conjunctiva ends in the caruncle, which contains both skin and conjunctival elements. Tenon’s capsule inserts into the bulbar conjunctiva and separates the conjunctiva from underlying tissues. Therefore, the bulbar and forniceal conjunctiva is only loosely attached to the posterior structures, including the levator and rectus muscle fascial tendon sheaths, allowing the motion of tissue upon eye and eyelid movement. The conjunctiva is also attached to the lower eyelid retractor muscles inferiorly and levator aponeurosis and Muller’s muscle superiorly. These attachments, in addition to the connections with the canthal ligaments, trochlea, and lacrimal gland, maintain the “suspensory apparatus” (2).
On a cellular level, the conjunctiva is made up of nonkeratinized, stratified columnar epithelium overlying the highly vascular connective tissue, the substantia propria. The columnar cells transition to stratified squamous epithelium continuous with the corneal epithelium (3). Goblet cells populate the conjunctiva in an inferonasal preponderance and are found in greater numbers in children. Limbal stem cells and mucocutaneous junction cells go on to form the bulbar and palpebral epithelial cells. In disease states, the destruction or damage of the limbal stem cells, may allow the conjunctival epithelium to grow onto the cornea creating a pannus.
Mucin, aqueous, and outer lipid layers comprise the tear film. Conjunctival goblet cells produce the mucopolysaccharides that, along with vesicles from the palpebral epithelial cells, form the mucous layer of the tear film (4). The lacrimal and accessory lacrimal gland ducts open to the conjunctival surface and produce the aqueous layer with its multitude of immune factors. The outer lipid layer is produced by the meibomian glands and Zeis’ glands. The lipid layer of the tear film is thicker in infants than in adults, with a longer tear breakup time (5). The proportion and interaction of these components create the stability of the tear film.
The conjunctiva protects the eye from pathogens by acting as a physical barrier and contributing immune components into the tear film. The immunologic function occurs in all layers of the conjunctiva: the superficial epithelial goblet cells creating immune protective mucin, the deeper epithelial basal layer infused with Langerhans’ cells, and the substantia propria containing mast cells, plasma cells, and neutrophils. The temporal lymphatic vessels transport lymph to the superficial parotid nodes while the nasal vessels drain into the submandibular nodes.
Bulbar innervation is derived from the ophthalmic division of the trigeminal nerve via the long ciliary nerves, branches of the nasociliary nerve. Superior palpebral and forniceal innervation is from the frontal and lacrimal branches of the ophthalmic division of the trigeminal nerve. Inferior palpebral and forniceal innervation is from the lacrimal branch of the ophthalmic division of the trigeminal nerve laterally and from the infraorbital nerve in the maxillary division of the trigeminal nerve.
The bulbar conjunctiva thins with age and loses elasticity as does the subconjunctival connective tissue or Tenon’s capsule. The posterior conjunctival arteries are smaller and
less tortuous in childhood. The combination of thinner, straighter blood vessels and a thicker Tenon’s capsule create the whiter appearance of young conjunctiva.
less tortuous in childhood. The combination of thinner, straighter blood vessels and a thicker Tenon’s capsule create the whiter appearance of young conjunctiva.
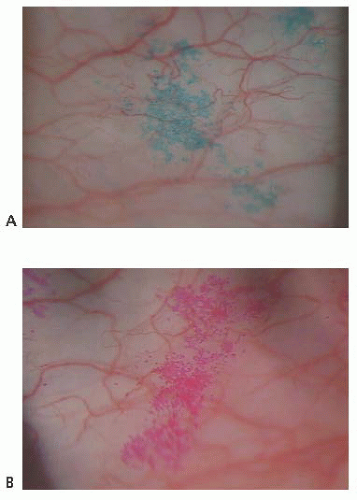 FIGURE 9.1. (A) Lissamine green versus rose bengal stain (B). |
Fluorescein is employed routinely to diagnose and differentiate inflammatory states as well as grossly quantitate the tear meniscus and breakup time. Rose bengal stain has been shown to stain damaged cells as well as cells with deficient mucous coating (6). Lissamine green dye stains dead or damaged epithelial cells, producing less stinging sensation than rose bengal (Fig. 9.1).
Pinguecula Pterygium and Actinic Episcleritis
Unlike adults, pinguecula and pterygium are rare in childhood. Children are subject to acute solar ultraviolet (UV) radiation however, which results in basophilic degeneration of the conjunctival substantia propria. Histologically, degeneration of the collagen fibers of the conjunctiva stroma with thinning of the overlying epithelium and occasional calcification occurs. Solar actinic exposure of the thin conjunctiva tissue results in fibroblasts producing more elastin fibers, but they are more twisted than normal, and may lead to the degradation of the collagen fibers. There is a marked decrease in normal elastin fibrils in these tissues (7). Clinically, these appear as a focal area of conjunctival vascular injection with underlying episcleritis (Fig. 9.2). At times, a fleshy elevated central area not contiguous with the limbus appears. Treatment includes lubricating drops and topical steroid.
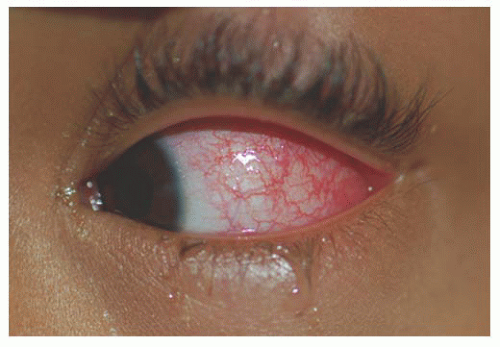 FIGURE 9.2. Actinic episcleritis with conjunctival vascular injection. |
Invasion of the cornea associated with Bowman’s layer destruction by a triangular area of bulbar conjunctiva (pterygium) can disrupt the tear film by creating an area of local drying on the cornea or dellen. Pterygia have increased numbers of mast cells (7). Pseudopterygia, which are usually not firmly adherent to the underlying tissue, can occur following corneal surgery or an inflammatory condition. Removal can be accomplished, as in adults, although recurrence is common.
PIGMENTATION AND PIGMENTED LESIONS
Conjunctival Icterus
Bilirubin has a high affinity for elastin, which is an abundant protein in the conjunctiva as well as the superficial, fibrovascular episclera, but not the sclera proper. An accurate description of the yellow jaundiced appearance of the eyes frequently seen transiently in neonates and chronically in children with hemolytic disorders is conjunctival icterus (8).
Melanocytic Nevi
Nevi are pigmented lesions classified like those of the skin: intraepithelial (junctional), subepithelial, compound (intraepithelial plus subepithelial), blue, and cellular blue (rarely seen in conjunctiva) (9). In adults, most conjunctival nevi are compound or subepithelial. Only in children are the pure intraepithelial (junctional) type observed (10).
Histologically, especially in young individuals, cytologic pleomorphism can be present in conjunctival nevi. A junctional nevus may be indistinguishable from primary acquired melanosis with atypia, a condition of elderly individuals that has a tendency to evolve into melanoma. Large spindle or epithelioid-shaped melanocytes characteristic of Spitz nevi may mimic melanoma (11). Inclusions of conjunctival epithelium in the form of solid islands or cysts are usually observed (12). During puberty, several changes can occur in the conjunctival nevi: the melanocytes may
proliferate or increase in pigmentation, and the epithelial cells within the inclusions may proliferate and secrete material, causing enlargement of the cysts. As the nevus becomes more prominent, it can cause irritation and inflammation. The inflammatory cell infiltrate can further increase the size, elevation, and vascularity of the nevus (Fig. 9.3). These alterations tend to provoke concern that a malignant melanoma has arisen from the nevus, resulting in excision of a large number of benign conjunctival nevi (9).
proliferate or increase in pigmentation, and the epithelial cells within the inclusions may proliferate and secrete material, causing enlargement of the cysts. As the nevus becomes more prominent, it can cause irritation and inflammation. The inflammatory cell infiltrate can further increase the size, elevation, and vascularity of the nevus (Fig. 9.3). These alterations tend to provoke concern that a malignant melanoma has arisen from the nevus, resulting in excision of a large number of benign conjunctival nevi (9).
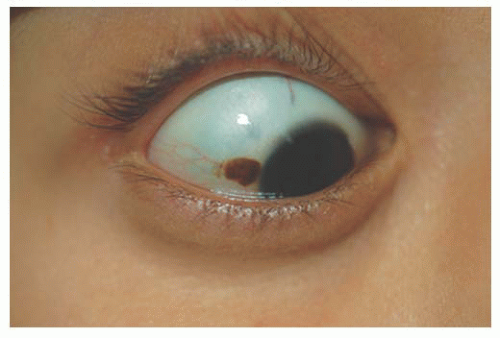 FIGURE 9.3. Nevus with typical inflammatory vascularization of conjunctiva. |
Nevus pigmentation is variable, with some devoid of pigment or amelanotic, requiring differentiation from other epithelial lesions. Occasionally, an inflamed nevus may become vascular and be mistaken for an angiomatous tumor (12).
Ocular Melanocytosis
Melanocytosis describes excessive melanotic pigmentation in the absence of a mass causing elevation of the conjunctiva that is typical of a nevus. Melanocytosis can be at the level of the conjunctiva or deeper in the sclera, choroid, or periocular tissues. Heterochromia may be present secondary to diffuse darkly pigmented micropapillae on the iris surface in place of iris crypts.
Epithelial congenital melanocytosis is a stationary lesion present at birth or early childhood. The lesions appear as irregular geographic patches of sclera and episcleral pigmentation that can range from distinct brown to gray in color. It is characterized by melanocytes and excessive melanin mainly in the basal layers of the conjunctival epithelium. It is not a precursor of malignant melanoma (9). Technically, the subepithelial congenital melanocytosis is not a lesion of the conjunctiva, because the abnormal melanocytes are found in the sclera and episclera. The overlying conjunctiva can be moved over the lesion without distorting it (13). There are two forms: (a) “ocular melanocytosis,” affecting ocular tissues only, and (b) “oculodermal melanocytosis,” or nevus of Ota, associated with ipsilateral melanosis of the lids or periocular facial skin. Most of the cases are unilateral and ipsilateral iris hyperpigmentation can be observed (14). It is more frequently seen in Asians and African descendants than in Caucasians (15,16) and may also be associated with melanosis of the orbital tissues and the meninges (10). Histologically, nevus of Ota is characterized by a congenital increase in the number, size, and pigmentation of the melanocytes of the uvea associated with increased numbers of pigmented melanocytes in the sclera, episclera, and dermis of the eyelids. Subepithelial congenital melanosis predisposes to the development of malignant melanoma (17,18). Children with oculodermal melanocytosis should be examined periodically because of a risk of pigmentary glaucoma and melanoma (19). Patients with ocular melanosis may also benefit from surveillance of the intraocular pressure (20) (Fig. 9.4). Some children with oculodermal melanocytosis may have pigmented lesions on the tongue and buccal mucosa (13).
Some syndromes and diseases may present with pigmented conjunctival lesions during their course, such as: (a) chronic forms of Gaucher’s disease; (b) alkaptonuria, in which the pigmentation can be seen over the horizontal recti insertion; (c) Kartagener’s syndrome, with characteristically marked conjunctival melanosis and hypertropia of the plica semilunaris; and (d) Peutz-Jegher’s syndrome, in which freckles can be seen over the conjunctiva, lids, and lips (21).
Gaucher’s Disease
Gaucher’s disease, found most commonly among Ashkenazic Jews, is a hepatorenal syndrome caused by enzyme deficiency of glucocerebrosidase, and is characterized by pinguecula-like lesions, corneal epithelial deposits, vitreous deposits, paramacular ring, white retinal infiltrates, oculomotor apraxia, hepatosplenomegaly, pancytopenia secondary to hypersplenism, bone pain, and accumulation of glucocerebroside. The pinguecula-like lesions can often be differentiated by their tan coloring and histologically contain Gaucher’s cells, enlarged lipid-laden macrophages. These lesions usually appear during the teenage years. The gene locus has been found on Chromosome 1.
 FIGURE 9.4. Ocular melanocytosis; congenital glaucoma. |
Alkaptonuria/Ochronosis
Alkaptonuria/ochronosis is a rare disorder of protein metabolism resulting in wasting of homogentisic acid in the urine. An enzyme deficiency of homogentisate-1,2-dioxygenase (ochronosis) results in the accumulation of homogentisic acid. The gene locus has been identified on chromosome 3q21-q23 and is inherited in an autosomal recessive fashion. This leads to pigment deposition in collagenous tissues. The manifestations are pigmented pinguecula, triangular scleral pigmentation near the horizontal rectus muscle insertions, episcleral pigment granules, oil-droplet opacities in the limbal corneal epithelium and Bowman’s layer, ochronotic arthropathy, ochronotic calculi in the genitourinary tract, and cardiovascular deposition, including calcification and stenosis in the aortic valve. Treatment is palliative.
Kartagener’s Syndrome
Kartagener’s syndrome was first described by Siewert in 1904 and later further characterized in the German literature by Kartagener in 1933. It is caused by mutations in the gene encoding the axonemal dynein intermediate chain. It is a rare syndrome associated with situs inversus (dextrocardia) and primary ciliary dyskinesia, leading to bronchiectasis and sinusitis. Conjunctival melanosis, hypertrophy of the semilunaris, myopia, and glaucoma have been described.
Peutz-Jegher’s Syndrome
Peutz-Jegher’s syndrome was initially described by Peutz, a Dutch internist, in 1921, as a familial syndrome. It is caused by mutations in the serine/threonine kinase STK11 gene chromosome locus 19p13.3. Peutz-Jegher’s syndrome is characterized by periocular and perioral melanocytic epidermal lesions (freckle-like appearance) in association with gastrointestinal polyps, usually of the small intestine. The macules present mainly on the lips and buccal mucosa but can appear on the palms, soles, and conjunctiva. The hyperpigmented macules precede the onset of gastrointestinal symptoms, including pain, bleeding, intussusception, and obstruction.
TUMORS AND INFILTRATES
Choristomas
Dermoids are the most common choristomas, lesions composed of tissue not normally found in the affected area. These solid, placoid tumors arise from the outer third of the sclera and often contain hair follicles, sebaceous glands, sweat glands, and fat lobules. Commonly occurring at the inferotemporal limbus, they appear as yellowish-white rounded elevations that are sometimes pigmented (Fig. 9.5). Most dermoids do not cause any discomfort but may produce significant astigmatism with secondary amblyopia. Ocular irritation may result from poor lid closure, tear film anomaly, or trauma from the fine hair that may grow from the surface of these lesions. Dermolipomas are more posterior and are associated with a large amount of fatty tissue. Usually, they arise near the insertion of the lateral rectus and are firmly fixed to the underlying sclera. Rarely, they can form symblepharontype adhesions, causing restriction of eye movement.
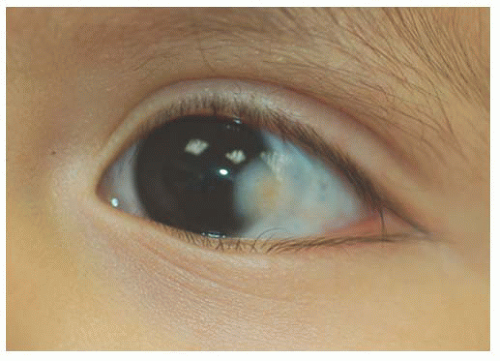 FIGURE 9.5. Limbal epibulbar dermoid choristoma. |
Dermoids and dermolipomas may be present with other systemic malformations, including Goldenhar’s syndrome (facio-auricular vertebral syndrome), mandibulofacial dysostosis (Treacher Collins syndrome, Franceschetti’s syndrome), and band-like cutaneous nevus and central nervous system dysfunction (Solomon’s syndrome, linear sebaceous nevus of Jadassohn). Both tend to grow with the patient and do not usually undergo neoplastic transformation.
Treatment is generally conservative. However, removal is indicated when significant astigmatism, with or without amblyopia, irritation, or cosmetic deformity, occurs. Lamellar excision is usually required because the outer third of the sclera is often involved. When the cornea is more involved, the surgeon should anticipate lamellar or penetrating keratoplasty. Complications of the excision include globe penetration, restriction to motility from scarring or injury of the associated rectus muscle, and increased astigmatism.
Other choristomas include ectopic lacrimal gland, simple and composed choristoma, and osseous choristoma. Ectopic lacrimal gland is the second most common choristoma affecting the epibulbar surface. Osseous choristomas are stationary lesions that resemble conjunctival dermoids; histologically, however, they are composed of mature compact bone surrounded by other choristomatous elements (22,23).
Children with the Organoid Nevus Syndrome present a birth with unilateral or bilateral epibulbar choristomas and raised linear or round lesions on the scalp containing sebaceous skin components (Fig. 9.6). Originally termed “nevus sebaceous of Jadassohn,” the skin lesions are now appropriately called organoid nevi because of the breadth of skin abnormalities they include. The most consistent ocular manifestations are epibulbar complex choristomas and posterior scleral cartilaginous choristomas. The epibulbar choristoma
is a fleshy pink lesion originating in the bulbar conjunctiva and may contain ectopic lacrimal gland tissue, fat cartilage, bone, nerve, and smooth muscle (Fig. 9.7). The choristoma may grow into the cornea in a progressive pattern after birth, causing partial or complete opacification (Fig. 9.8). These may prove difficult to excise (24).
is a fleshy pink lesion originating in the bulbar conjunctiva and may contain ectopic lacrimal gland tissue, fat cartilage, bone, nerve, and smooth muscle (Fig. 9.7). The choristoma may grow into the cornea in a progressive pattern after birth, causing partial or complete opacification (Fig. 9.8). These may prove difficult to excise (24).
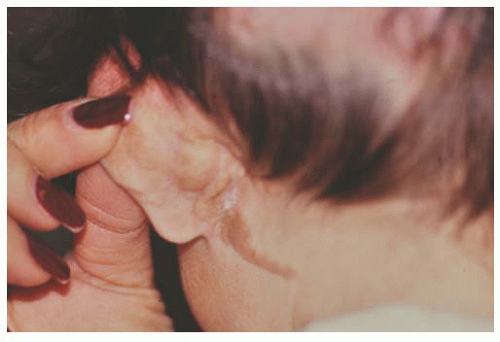 FIGURE 9.6. Posterior scalp and ear of child with organoid nevus syndrome with linear sebaceous nevus. |
Hamartomas
Hamartomas are lesions composed of tissue found normally in the affected area. Neurofibromas are solid nodular lesions affecting the bulbar or palpebral conjunctiva. They can be plexiform, solitary, or diffuse type and are almost always associated with neurofibromatosis type 1 or type 2. Type 2B multiple endocrine neoplasia (MEN) is associated with medullary thyroid carcinoma and pheochromocytoma. Ocular abnormalities include mucosal neuroma located on the conjunctiva at the limbus and on the palpebral conjunctiva proximal to the eyelid margin. Prominent medullated corneal nerves occur in 100% of cases. These patients have a marfanoid habitus including the findings of a high-arched palate, pectus excavatum, bilateral pes cavus, and scoliosis. Neuromas on the eyelids, conjunctiva, nasal and laryngeal mucosa, tongue, and lips are frequent findings. The conjunctival neuromas and neurofibromas appear as yellow-gray sessile or dome-shaped masses located in the stroma. Patients also have prominent, hypertrophied lips leading to a characteristic facial appearance. The male-to-female ratio for MEN is 2:1. The disease may appear in children younger than 10 years. However, all MEN syndromes are rare in children (13).
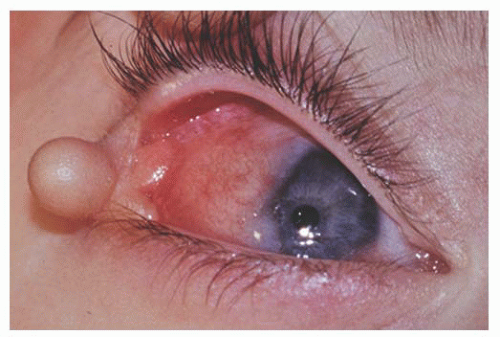 FIGURE 9.7. Epibulbar complex choristoma with skin pedicle containing bone and cartilage in organoid nevus syndrome. |
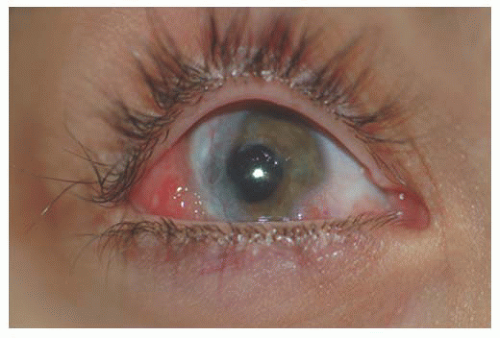 FIGURE 9.8. Progressive infiltration of the cornea by choristoma in organoid nevus syndrome. |
Fibrous hamartomas are epibulbar lesions that contain abundant mature elastic fibers intermixed with fibrous tissue. They may be seen in patients with Proteus syndrome, a rare syndrome characterized by asymmetrical overgrowth that can affect any structure (bones, skin, viscera) and is generally progressive throughout childhood.
Another hamartomatous lesion that affects the conjunctiva is hemorrhagic lymphangiectasia. These lesions are irregularly dilated lymphatic channels of the bulbar conjunctiva that may sometimes be filled with blood. They may arise as a developmental anomaly or in association with trauma or inflammation (22,23).
Conjunctival Cysts
Conjunctival cysts are stable lesions that can be congenital or acquired. A common cause of acquired conjunctival inclusion cysts is the implantation of conjunctival epithelium islands after surgery or trauma. They may disappear spontaneously, but persistent cases often require surgical excision or diathermy (22).
Pyogenic Granuloma
Pyogenic granuloma is a vasoproliferative inflammatory response composed of granulation tissue. The term “pyogenic granuloma” is a misnomer. The lesion neither causes pus formation nor is a true granuloma. The pathogenesis is
not well understood, but they can develop rapidly and are often associated with previous ocular and adnexal surgery, inflammation, foreign bodies, chemical burns, or phthisis bulbi. Following strabismus surgery these lesions may occur near the sutured margin of the conjunctiva. Spontaneous involution can occur or, if necessary, simple excisional biopsy with cautery of the base can be both diagnostic and curative (22) (Fig. 9.9).
not well understood, but they can develop rapidly and are often associated with previous ocular and adnexal surgery, inflammation, foreign bodies, chemical burns, or phthisis bulbi. Following strabismus surgery these lesions may occur near the sutured margin of the conjunctiva. Spontaneous involution can occur or, if necessary, simple excisional biopsy with cautery of the base can be both diagnostic and curative (22) (Fig. 9.9).
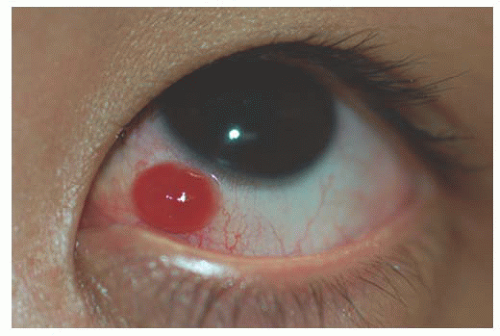 FIGURE 9.9. Post strabismus surgery pyogenic granuloma. |
Xeroderma Pigmentosa Syndrome
This disease is a rare autosomal recessive disorder that manifests as an inability to repair deoxyribonucleic acid damage induced by UV radiation. Clinically, this syndrome is characterized by the early development of pigmentary changes, atrophy, keratoses, and skin malignancies (carcinomas, melanomas, sarcomas, angiosarcomas, neuromas), predominantly on light-exposed skin. Some patients may present with recurrent conjunctivitis, dry eyes with areas of pigment deposition and keratin formation, as well as squamous cell carcinomas of the conjunctiva. The syndrome is sometimes associated with slowly progressive neurologic abnormalities that include deafness, ataxia, mental retardation, and cerebellar atrophy. Most patients die of malignancy before age 20 (25). Treatment is avoidance of sunlight and UV exposure by use of sunblock and 100% UV-barrier spectacles with sidearms (23).
Benign Hereditary Epithelial Dyskeratosis
This is a rare disorder that affects primarily members of the Haliwa Indians (Halifax and Washington counties of North Carolina). It is an autosomal dominant disorder with high penetrance characterized by bilateral elevated plaques in the exposed areas of the conjunctiva. Benign hereditary epithelial dyskeratosis has a chronic course of ocular irritation/photophobia and may be associated with dyskeratosis of the oral mucosa. Histologically, the lesions show epithelial acanthosis, parakeratosis, and dyskeratosis, with the stroma usually containing chronic inflammatory cells. Atypia is absent, and there is no dysplastic potential. Treatment of choice is excision of the lesions, although the recurrence rate is high (22,23).
VASCULAR ABNORMALITIES
Hemangiomas are lesions composed predominantly of blood vessels and may occur as isolated lesions or in association with lid, orbital, or intracranial lesions. Clinically, they appear as red masses on the conjunctival surface and blanch on pressure. Spontaneous hemorrhage is not infrequent and may occur after trivial trauma. Surgical excision is difficult, and recurrences may occur. The surface vasculature in capillary hemangiomas involutes following the application of topical timolol 0.5% in some cases (26).
Lymphangiomas are usually widespread, occasionally affecting an entire hemiface. Clinically, these lesions show clear fluid-filled cystic areas among the blood-filled hemangioma tissue (Fig. 9.10). Surgical excision is difficult due to the diffuse nature of the lesions (13).
Several syndromes can have conjunctival vascular abnormalities among their features. The characteristic feature of Sturge-Weber syndrome is a cutaneous hemangioma, commonly in a trigeminal facial distribution, with the conjunctiva showing only a faint blush on the normal whiteness of the conjunctiva. Klippel-Trenaunay-Weber syndrome, a widespread vascular anomaly causing limb hypertrophy and vascular anomalies of skin, may also have conjunctival angiomas. Conjunctival vascular malformations may be seen associated with racemose angiomatous malformation of the retina in Wyburn-Mason syndrome. Louis-Bar syndrome or ataxia telangiectasia (AT) is an autosomal recessive disorder that presents with a progressive ataxia and degeneration of central nervous system function (choreoathetosis, dysrhythmic speech, aberrant ocular movements and occasional seizures) and presence of extremely tortuous and telangiectatic conjunctival vessels without an associated
lymphatic component in exposed areas of conjunctiva and skin (Fig. 9.11). In Rendu-Osler-Weber syndrome, conjunctival telangiectases may be seen along with retinal vascular malformation (13).
lymphatic component in exposed areas of conjunctiva and skin (Fig. 9.11). In Rendu-Osler-Weber syndrome, conjunctival telangiectases may be seen along with retinal vascular malformation (13).
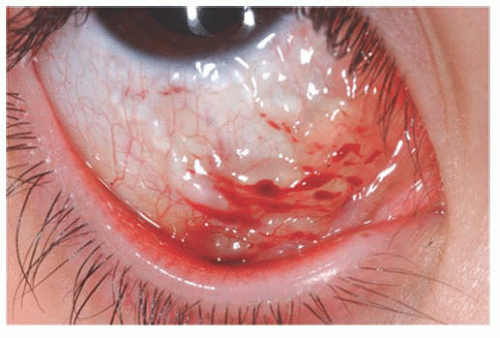 FIGURE 9.10. Lymphangioma with clear fluid-filled cystic areas among the blood-filled hemangioma tissue. |
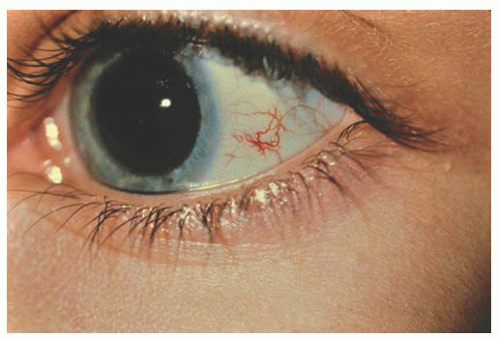 FIGURE 9.11. Tortuous and telangiectatic conjunctival vessels in ataxia telangiectasia (AT). |
CONJUNCTIVITIS OF THE NEWBORN
Neonatal conjunctivitis is inflammation of the conjunctiva that affects infants during the first month after birth. It is usually a hyperacute papillary conjunctivitis, because a follicular response is not seen before 6 to 8 weeks of life (27).
The period of time after birth until the onset of the conjunctivitis is variable, but it may be helpful in suggesting the cause by correlating it to the incubation time of the possible etiologic agents.
When the conjunctivitis affects the baby in the first few days after birth, the probable cause is the toxic effect of the prophylactic agent used at birth. It is characterized by mild and transient conjunctival injection and tearing, which usually resolves in 24 to 48 hours. This was more common when silver nitrate was used for prophylaxis.
Conjunctivitis due to Neisseria gonorrhoeae typically appears 2 to 5 days after birth. It is still common in developing countries but rare in the most industrialized countries. Clinically, it starts with a serosanguineous discharge that rapidly progresses to a thick purulent discharge associated with markedly edematous eyelids and chemosis. Conjunctival membranes may be seen. This bacteria has the propensity to produce a severe keratitis because of its ability to penetrate intact epithelial cells and replicate rapidly. A delay in diagnosis and treatment may lead to corneal ulceration and perforation.
Historically, the latent period for bacterial conjunctivitis, other than N. gonorrhoeae has been 5 to 8 days after birth, but it can occur any time in the immediate postpartum period. Etiologic agents include: Haemophilus sp., Streptococcus pneumoniae, Staphylococcus aureus, and rarely, Pseudomonas aeruginosa. Although rare, this bacteria can rapidly progress from conjunctivitis to corneal ulceration and perforation. If Pseudomonas is not recognized as the etiologic agent, the conjunctival infection may lead to endophthalmitis and possible death.
Chlamydia trachomatis serotype D-K, causing neonatal inclusion conjunctivitis represents the most common isolated pathogen in newborns with conjunctivitis in industrialized countries. The incubation period ranges from 5 to 14 days. Clinically, one sees a mild mucopurulent conjunctivitis associated with moderate lid swelling and mild chemosis typically beginning as unilateral but often becoming bilateral (Fig. 9.12). Although generally considered benign and self-limited with spontaneous resolution in 8 to 12 months, if untreated it may result in the formation of a micropannus and scarring of the tarsal conjunctiva. Systemic spread of this ocular infection can cause disease involving the pharynx, lungs, and/or rectum, which can be fatal. The systemic potential of this disease dictates the need for systemic treatment, generally with a macrolide antibiotic.
Viral neonatal conjunctivitis caused by Herpes simplex virus (HSV) typically occurs within 6 to 14 days afterbirth. Although 80% of the babies affected with HSV have typical herpetic lesions on the skin, eyelids, or mouth, without these lesions the conjunctivitis is indistinguishable from other causes of neonatal conjunctivitis. Signs of corneal involvement include microdendrites or geographical ulcers. Herpetic keratoconjunctivitis is frequently associated with systemic infection, with mortality rates of disseminated disease around 50%.
A less frequent cause of neonatal conjunctivitis is Candida albicans. It presents as a pseudomembranous conjunctivitis, with the average time of onset of 5 days after exposure.
Congenital nasolacrimal duct obstruction is also frequently associated with conjunctivitis of the newborn typically caused by Haemophilus sp. and S. pneumoniae.
Clinically, these differentiation among the various etiologies can be difficult. However, because of the potentially serious complications of an improperly treated conjunctivitis, determination of the causative agent is recommended
at this age. Conjunctival smears and appropriate cultures should be done in all patients. Scrapings of the conjunctiva should be collected with a spatula and cultures obtained with a calcium-alginate swab, prewetted with sterile liquid culture media. Gram and Giemsa stains should be performed on the conjunctival scraping. Giemsa stain in chemical conjunctivitis shows neutrophils with occasional lymphocytes; in chlamydial infection, neutrophils, lymphocytes, plasma cells, and basophilic intracytoplasmic inclusions in epithelial cells; in viral conjunctivitis, lymphocytes, plasma cells, multinucleated giant cells, and eosinophilic intranuclear inclusions; in fungal infections, neutrophils, and pseudohyphal budding yeast formation; and in bacterial infections, neutrophils, and bacteria. Certain bacteria can be identified on Gram stain. Gram-negative diplococci with polymorphonuclear leucocytes suggest gonococcal infection; Gram-negative coccobacilli correlate with Haemophilus sp.; Gram-positive cocci suggest S. aureus and S. pneumoniae. Recommended medias for culture include: reduced blood agar, thioglycolate, brainheart infusion broth for aerobic bacteria; chocolate agar in CO2 or Thayer-Martin for N. gonorrhoeae; Sabouraud’s slant for fungus. McCoy cell culture has been the standard for diagnosing C. trachomatis in the past, but this technique is expensive and requires at least 2 to 3 days for results. Polymerase chain reaction (PCR) analysis and direct immunofluorescent monoclonal antibody stain (DFA) have comparable specificity with higher sensitivities and faster results than traditional culture testing. Viral cultures are expensive and take 2 to 4 days to grow. Viral antigens can be detected rapidly, using immunologic tests, such as direct immunofluorescent testing, enzyme-linked immunosorbent assay (ELISA), and immunofiltration method.
at this age. Conjunctival smears and appropriate cultures should be done in all patients. Scrapings of the conjunctiva should be collected with a spatula and cultures obtained with a calcium-alginate swab, prewetted with sterile liquid culture media. Gram and Giemsa stains should be performed on the conjunctival scraping. Giemsa stain in chemical conjunctivitis shows neutrophils with occasional lymphocytes; in chlamydial infection, neutrophils, lymphocytes, plasma cells, and basophilic intracytoplasmic inclusions in epithelial cells; in viral conjunctivitis, lymphocytes, plasma cells, multinucleated giant cells, and eosinophilic intranuclear inclusions; in fungal infections, neutrophils, and pseudohyphal budding yeast formation; and in bacterial infections, neutrophils, and bacteria. Certain bacteria can be identified on Gram stain. Gram-negative diplococci with polymorphonuclear leucocytes suggest gonococcal infection; Gram-negative coccobacilli correlate with Haemophilus sp.; Gram-positive cocci suggest S. aureus and S. pneumoniae. Recommended medias for culture include: reduced blood agar, thioglycolate, brainheart infusion broth for aerobic bacteria; chocolate agar in CO2 or Thayer-Martin for N. gonorrhoeae; Sabouraud’s slant for fungus. McCoy cell culture has been the standard for diagnosing C. trachomatis in the past, but this technique is expensive and requires at least 2 to 3 days for results. Polymerase chain reaction (PCR) analysis and direct immunofluorescent monoclonal antibody stain (DFA) have comparable specificity with higher sensitivities and faster results than traditional culture testing. Viral cultures are expensive and take 2 to 4 days to grow. Viral antigens can be detected rapidly, using immunologic tests, such as direct immunofluorescent testing, enzyme-linked immunosorbent assay (ELISA), and immunofiltration method.
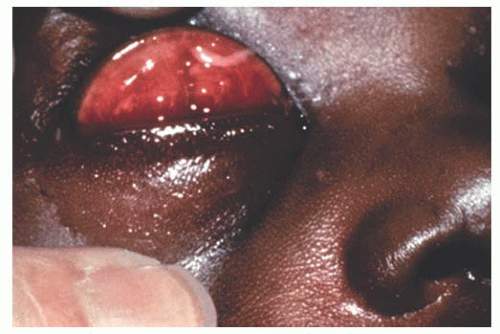 FIGURE 9.12. Neonatal inclusion conjunctivitis caused by Chlamydia trachomatis with pseudomembrane formation. |
Treatment is not required for chemical conjunctivitis caused by prophylactic agents, which typically resolves spontaneously in 24 to 48 hours. According to the World Health Organization (WHO) guidelines, all cases of conjunctivitis in the newborn should be treated for both N. gonorrhoeae and C. trachomatis because of the possibility of mixed infection.
The treatment of the N. gonorrhoeae conjunctivitis consists of intravenous Penicillin G 100,000 units/kg/day for 7 days. N. gonorrhoeae resistant to penicillin is found in many urban areas in the United States and worldwide. In this case, the conjunctivitis should be treated with a third-generation cephalosporin. A single intramuscular dose of ceftriaxone 125 mg is highly effective and is the recommended treatment by WHO guidelines (28,29




Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
