Congenital tracheal lesions are rare, but important, causes of morbidity in infants and children. Consequently, experience in their management is limited and dispersed. Given its small diameter, the juvenile trachea is obstructed easily by various natural causes, or following a surgical intervention. The diagnosis of a congenital, tracheal, obstructive anomaly is based on a high degree of suspicion in infants and children with respiratory distress accompanied by retraction. In this article, the authors discuss the various causes of these conditions, their diagnostic features, and the treatment possibilities.
Congenital tracheal lesions are rare, but important, causes of morbidity in infants and children. Consequently, experience in their management is limited and dispersed. Given its small diameter, the juvenile trachea is obstructed easily by various natural causes, or following a surgical intervention. One must be aware of the various causes of these conditions, their diagnostic features, and the treatment possibilities.
Embryology of the trachea
The laryngotracheal groove, or the sulcus, appears in the proximal foregut at the third week (3 mm embryo, stage10). This groove slowly progresses caudad, and the lateral ridges form the primordium of the trachea. The pulmonary primordium appears and bulges ventrally from the foregut. Complete separation of trachea and esophagus occurs by the 11 to 14 mm stage (sixth week). The tip of the tracheal primordium buds asymmetrically, left and right, at the 4 mm stage, to provide the bronchial primordia. Mesenchymal proliferation by cells lining the coelomic cavity provides the tissue from which cartilage, muscle, and connective tissue develops. Epithelial–mesenchymal interrelationships are essential for bronchial and pulmonary development to occur. The tracheal bifurcation moves downward gradually from the neck to the level of the fourth vertebra. Cartilage appears in the trachea at 10 weeks.
Failure of complete separation of the foregut into respiratory and alimentary components is the most common defect, and produces tracheoesophageal fistula (TEF). At the upper end, the larynx may fail to reopen, producing atresia (usually a fatal anomaly), or it may fail to form a complete posterior septum, producing a laryngotracheoesophageal cleft (LTEC). Tracheal atresia, stenosis, and TEF may occur more distally. The relatively separate processes of laryngeal development, and budding of bronchi and pulmonary development, allow for malformations of the trachea, such as agenesis and stenosis, in the presence of a normal larynx and bronchial tree.
Anatomy
The trachea occupies the anterior and middle part of the neck and penetrates into the superior mediastinum behind the sternum. It begins at the level of the cricoid cartilage and ends at the level of the sternal angle, where it divides to form the two main or, primary, bronchi. The anterior end of the trachea is at the level of the fifth thoracic vertebra, or the sternal angle. The trachea is 4 cm long in a full-term newborn infant, and 11 to 13 cm long in an adult. The diameter of the trachea is 3 to 4 mm in a full-term newborn, and 12 to 23 mm in an adult. The lateral and anterior walls of the trachea are supported by 16 to 20 horseshoe-shaped cartilages . Tracheal cartilages are C-shaped hyaline cartilages. The posterior or membranous portion of the trachea is composed of the trachealis muscle, and elastic and fibrous tissue. The ratio of cartilaginous to membranous trachea normally is 4.5:1; however, this tracheal cross-section changes during breathing and coughing as a result of changes in head and neck position and intrathoracic pressure.
The trachea has two depressions: a superior depression formed by the left thyroid lobule, and an inferior depression near the bifurcation made by the aorta. The lumen of the trachea is lined by a mucous membrane consisting of a thin lamina propria and ciliated, pseudostratified columnar epithelium. The superior and inferior thyroid, thymic, and right bronchial arteries provide the arterial supply of the trachea. The veins form rings that travel along the intercartilaginous spaces and flow into the esophageal and inferior thyroid veins. Tracheal innervation comes from the vagus nerve (pulmonary plexus and laryngeal nerves) and the sympathetic nerves (cervical and dorsal ganglia).
Assessment
Clinical findings
The diagnosis of a congenital, tracheal, obstructive anomaly is based on a high degree of suspicion in infants and children with respiratory distress accompanied by retraction. The child may have a history of respiratory difficulties of lesser intensity since birth, or shortly after birth, or of repeated and stubborn respiratory infections. Strangely, dyspnea may be episodic. Cyanosis and apneic episodes may occur. In some cases, difficulty in intubation leads to the diagnosis. Often, recurrent or persistent cough and exercise intolerance are associated. See later discussion for characteristic features of each condition.
Endoscopy
Careful use of a flexible pediatric bronchoscope can clarify much about a lesion. Rigid bronchoscopy is a procedure that allows safe ventilation during inspection, but it has to be perfectly atraumatic. The bronchoscope should not be passed into a tightly stenotic lesion, to avoid edema and inflammation, which might precipitate acute obstruction. Rigid ventilating pediatric bronchoscopes can be used for bronchoscopic evaluation. However, either a flexible pediatric bronchoscope or a long telescope (outer diameter of 2 mm) allows a more distal examination. These may be inserted through a larger, rigid, ventilating bronchoscope seated proximally, or through a pediatric operating laryngoscope. Bronchoscopes should not be forced into a stenosis, nor should any attempt be made to dilate a congenital narrowing. Ventilation is maintained by the intermittent placement of an endotracheal tube or supraglottic jet ventilation when using the laryngoscope alone for exposure. The esophagoscopy demonstrates esophageal malformations or external (often vascular) compressions. TEFs are found in the membranous wall of the trachea. Instillation of methylene blue saline into the esophagus by way of a high-placed nasogastric tube may identify a small H-type fistula conclusively.
Imaging
CT and MRI provide precise information about the cross-sectional area and extent of lesions, with three-dimensional reconstruction available. They also help in a complete mediastinal and pulmonary evaluation, which may aid in the diagnosis of associated congenital malformations. A spiral CT, because of its short duration, does not require sedation in infants. MRI offers similar information and is useful especially to delineate associated cardiovascular anomalies. Echocardiography allows a good cardiovascular evaluation, but it is often complemented by cardiac catheterization in those patients who are to undergo surgery. Angiography is also used less often, but is still the “gold standard” for precise and complete delineation of vascular anomalies .
Tracheal agenesis/atresia
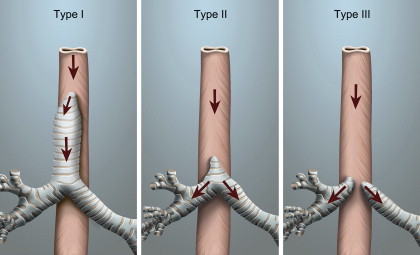
Tracheal agenesis is a rare congenital anomaly that is not compatible with prolonged life, even with current medical technology. Its incidence is reported to be less than 1:50,000, with a male predominance . The disorder was first described in 1900 by Payne, after the death of an infant on whom a tracheotomy had been attempted . The absence of a trachea was noted on postmortem examination. Diagnosis can be made if the neonate presents with respiratory distress at birth, is unable to produce an audible cry in spite of an obvious physical effort, and cannot be intubated but shows some improvement when ventilated by bag and mask. An accidental esophageal intubation may improve the respiratory status temporarily if a TEF is present. In 1962, Floyd proposed an anatomic classification of this malformation ( Fig. 1 ). Type I is atresia of part of the trachea with a normal but short distal trachea, normal bronchi, and a TEF. This type makes up approximately 20% of the malformations. Sixty percent of the reported cases are type II, where there is complete tracheal atresia but normal bifurcation and bronchi. Type III, accounting for 20% of cases, has no trachea and the bronchi arise directly from the esophagus .
Kluth and colleagues showed that the esophagus and trachea develop as the foregut decreases in size by infolding without formation of a fused septum. They proposed that tracheal atresia with fistula may result from a ventral deformation of the foregut and a concomitant dorsal dislocation of the tracheoesophageal space. Baarsma and colleagues suggested that this condition should be suspected if the antenatal ultrasound examination shows abnormal fetal breathing movement. Antenatal ultrasonography may show bilateral uniform hyperechoic lungs and ascites if the trachea or larynx is obstructed completely. The inspissated lung secretions cause overdistension of the lungs. Inversion or flattening of the diaphragm can occur. Contiguous compression of the fetal heart results in low-output heart failure. In the presence of an esophageal fistula, the lungs do not become enlarged because fluid escapes through the fistula into the gastrointestinal tract. Color flow Doppler ultrasonographic findings may show an absence of blood flow at the laryngeal level . The most common variant has normal bronchi communicating centrally to the esophagus.
Other congenital anomalies are common in these patients. Fonkalsrud and colleagues described a newborn who survived for a short time by using the esophagus as an airway . A major bronchus may also communicate directly with the esophagus while the balance of the lung is served by anomalous bronchi from a partly stenotic trachea. Microgastria is a common concomitant feature. A systematic surgical approach to these anomalies does not exist, doubtless because of their rarity and variability, and the complexity of the defect . Hiyama and colleagues treated one of their two infant patients successfully with the following procedures: gastrostomy and abdominal esophageal banding, translaryngeal and esophageal ventilation by endotracheal tube, tracheostomy and later T tube, pharyngeal sump drainage followed by establishment of cervical esophagostomy (proximal tracheal segment present), and esophageal reconstruction by colonic interposition at age 3 years .
The survival of infants with tracheal agenesis is rare, and correction is very difficult. Short-term survival may be possible if there is a fistulous connection between the esophagus and bronchus. So far, 11 cases with varying degrees of tracheal agenesis have been resuscitated and treated with various palliative and tracheal reconstructive procedures. Published reports show long-term survival of three of them . Attempts at using the esophagus as an air conduit are only temporary, but may allow sufficient time for the diagnosis to be confirmed. It is hoped that the use of tissue-engineered cartilage may improve the outcome of affected babies without other major associated congenital anomalies.
Tracheal agenesis/atresia
Tracheal agenesis is a rare congenital anomaly that is not compatible with prolonged life, even with current medical technology. Its incidence is reported to be less than 1:50,000, with a male predominance . The disorder was first described in 1900 by Payne, after the death of an infant on whom a tracheotomy had been attempted . The absence of a trachea was noted on postmortem examination. Diagnosis can be made if the neonate presents with respiratory distress at birth, is unable to produce an audible cry in spite of an obvious physical effort, and cannot be intubated but shows some improvement when ventilated by bag and mask. An accidental esophageal intubation may improve the respiratory status temporarily if a TEF is present. In 1962, Floyd proposed an anatomic classification of this malformation ( Fig. 1 ). Type I is atresia of part of the trachea with a normal but short distal trachea, normal bronchi, and a TEF. This type makes up approximately 20% of the malformations. Sixty percent of the reported cases are type II, where there is complete tracheal atresia but normal bifurcation and bronchi. Type III, accounting for 20% of cases, has no trachea and the bronchi arise directly from the esophagus .
Kluth and colleagues showed that the esophagus and trachea develop as the foregut decreases in size by infolding without formation of a fused septum. They proposed that tracheal atresia with fistula may result from a ventral deformation of the foregut and a concomitant dorsal dislocation of the tracheoesophageal space. Baarsma and colleagues suggested that this condition should be suspected if the antenatal ultrasound examination shows abnormal fetal breathing movement. Antenatal ultrasonography may show bilateral uniform hyperechoic lungs and ascites if the trachea or larynx is obstructed completely. The inspissated lung secretions cause overdistension of the lungs. Inversion or flattening of the diaphragm can occur. Contiguous compression of the fetal heart results in low-output heart failure. In the presence of an esophageal fistula, the lungs do not become enlarged because fluid escapes through the fistula into the gastrointestinal tract. Color flow Doppler ultrasonographic findings may show an absence of blood flow at the laryngeal level . The most common variant has normal bronchi communicating centrally to the esophagus.
Other congenital anomalies are common in these patients. Fonkalsrud and colleagues described a newborn who survived for a short time by using the esophagus as an airway . A major bronchus may also communicate directly with the esophagus while the balance of the lung is served by anomalous bronchi from a partly stenotic trachea. Microgastria is a common concomitant feature. A systematic surgical approach to these anomalies does not exist, doubtless because of their rarity and variability, and the complexity of the defect . Hiyama and colleagues treated one of their two infant patients successfully with the following procedures: gastrostomy and abdominal esophageal banding, translaryngeal and esophageal ventilation by endotracheal tube, tracheostomy and later T tube, pharyngeal sump drainage followed by establishment of cervical esophagostomy (proximal tracheal segment present), and esophageal reconstruction by colonic interposition at age 3 years .
The survival of infants with tracheal agenesis is rare, and correction is very difficult. Short-term survival may be possible if there is a fistulous connection between the esophagus and bronchus. So far, 11 cases with varying degrees of tracheal agenesis have been resuscitated and treated with various palliative and tracheal reconstructive procedures. Published reports show long-term survival of three of them . Attempts at using the esophagus as an air conduit are only temporary, but may allow sufficient time for the diagnosis to be confirmed. It is hoped that the use of tissue-engineered cartilage may improve the outcome of affected babies without other major associated congenital anomalies.
Tracheobronchomalacia
In the pediatric age group, primary or secondary tracheobronchomalacia (TBM) is the most common cause of significant airway collapse with resultant obstruction. These problems often present difficult management issues. Various underlying pathologies may lead to increased resistance in air flow and hence, increased work of breathing. Mucociliary clearance can also be impaired causing various sequelae. In most children with TBM, the condition is self-limited, with resolution occurring within 1 to 2 years. Patients who have dying spells, recurrent pneumonia, or those who cannot be weaned from long-term ventilation may require more permanent management. TBM is a rare condition of infancy and childhood, characterized by abnormal compliance of the tracheobronchial tree . Occasionally, TBM is an isolated lesion (primary TBM). More commonly, however, the condition is associated with other congenital abnormalities ( Box 1 , Figs. 2 and 3 ), including esophageal atresia and TEF. TBM may also be secondary to extrinsic compression from vascular structures or soft tissue masses. Rarely, a primary disorder of collagen-like dyschondroplasia or polychondritis may lead to TBM.
1. Esophageal atresia with TEF
2. Extrinsic compression
Vascular causes (innominate artery, aortic arch ring, pulmonary artery sling, aberrant right subclavian)
Cardiac causes (enlarged left atrium, enlarged pulmonary arteries, enlarged pulmonary veins)
Cysts (lymphatic malformations, thymic cysts, bronchogenic cysts)
Mediastinal neoplasm (teratoma, lymphoma, neuroblastoma)
Infection (abscess)
3. Prolonged Intubation
4. Chondrodysplasias
5. Posttraumatic result (or following tracheoplasty)
Pathophysiology
In laminar airflow, resistance is inversely proportional to the fourth power of the radius. Thus, even small changes in airway caliber can have dramatic effects on airway resistance. In a normal airway, a stiff cartilaginous framework maintains patency, despite changing intrathoracic pressures during the respiratory cycle. During expiration, positive intrathoracic pressure is transmitted to the intrathoracic airways and causes a physiologic narrowing. In patients who have TBM, the abnormally compliant tracheobronchial cartilages are not able to resist increased intrathoracic pressure, leading to a collapse of the airway in the anteroposterior direction. This condition is aggravated during forced expiration and coughing, and during the Valsalva’s maneuver, where the intrathoracic pressures are even higher. Also, compression by surrounding structures, anteriorly by the aorta and posteriorly by the esophagus, adds to airway collapsibility.
Pathology
In TBM patients, the tracheal cartilage is deficient or malformed, with a decreased ratio of cartilage to muscle ( Fig. 4 ). Pathologic examination of major malacic segments reveals a decrease in the amount, size, and thickness of cartilaginous plates. Immaturity of major airway cartilage is thought to cause primary TBM. Secondary TBM likely occurs to some degree in all children with esophageal atresia. It has been postulated that an in utero tracheal compression forms a dilated esophageal pouch, leading to abnormal development. An associated loss of intratracheal pressure through a TEF may cause an additional collapsibility. Extrinsic tracheal compression is associated with tracheal cartilage abnormalities, such as hypoplasia, dysplasia, or a deficiency in the normal cartilaginous framework. Usually, a reduction in the amount of tracheal cartilage is combined with an increased width of membranous trachea in patients who have TBM. This combination of shortened, weaker tracheal rings and wider, posterior, membranous trachea results in a loss of the classic (cross-sectional) “D” configuration of the trachea. The trachea becomes compressed in the anteroposterior plane, and, in some segments, weakened cartilage is fractured into two to four pieces.
Signs and symptoms
Symptoms of TBM can manifest at birth, but often are evident only after 2 months of age. A brassy cough and expiratory stridor are the most common symptoms. This cough likely is caused by the abutment of the anterior and posterior walls of the trachea, which results in vibrations during cough. The most serious symptoms are dying spells or death attacks. These spells usually occur during feeding, or within 10 minutes of a meal. Patients become cyanotic and apneic, and often lose general muscle tone, which is believed to be caused by esophageal dilatation by a food bolus compressing the malacic segment from behind. If the feeding is not stopped, the symptoms can progress, leading to cardiorespiratory arrest. Respiratory infections are seen commonly in children with TBM. Respiratory secretions accumulate in distal airways because of airway collapse at the end of expiration, and may cause recurrent pneumonias.
Treatment options
In many children with TBM, intervention is not necessary. As the child grows, the tracheal cartilage strengthens and stiffens, and the symptoms often resolve by age 1 to 2 years in children with mild-to-moderate TBM. Therefore, conservative therapy in milder cases is preferred, and includes the treatment of respiratory infections, humidified oxygen therapy, and pulmonary physiotherapy. For children who do not recover spontaneously, or who have life-threatening symptoms, various treatment options are available.
The treatment of TBM has advanced in the last 3 decades. In the past, the mainstay of therapy in patients who had severe TBM was tracheotomy and long-term mechanical ventilation. Pioneering physicians recognized early that standard tracheotomy tubes were not always long enough to stent the involved segment of the trachea. Physicians developed elongated tracheotomy tubes, or advanced thin-walled tubes through a standard tracheotomy, to mechanically stent the distal trachea. The problems with this approach included the need to change the length of the tube as the child grew, recurrent bronchospasm, the negation of the glottic mechanism of increasing intratracheal pressure, and difficult decannulation. In addition, chronic tracheotomy produces some tracheal injury and predisposes one to recurrent infections, which has spurred the search for alternatives. The percentage of infants and children who still require tracheotomy for TBM varies from 12% to 62% . Jacobs and colleagues reviewed 50 cases of TBM, and reported that tracheotomy was required in 75% of premature infants and in 39% of full-term infants. Of those, 71% were able to undergo decannulation after an average of 30 months, without further intervention.
Continuous positive airway pressure (CPAP) is an effective treatment for infants with moderate to severe TBM . Bronchoscopy and fluoroscopy have shown that CPAP maintains airway patency during tidal breathing. By creating a “pneumatic stent,” CPAP prevents the collapse of the airway throughout the respiratory cycle. Tidal-breathing, flow-volume measurements have been used to evaluate the changes in airway obstruction with the use of CPAP. However, CPAP has some disadvantages , including a lag in the commencement of oral feedings, retardation of speech and language, and potential developmental delay. Depending on the estimated duration of treatment, CPAP can be considered a primary treatment, or an adjuvant to other therapies. In patients who have the most severe forms of TBM, or who present with life-threatening symptoms, surgical intervention may be necessary. Indications for surgery include recurrent pneumonia, intermittent respiratory obstruction, and the inability to extubate the airway . Dying spells are an indication for continued hospitalization and definitive surgical intervention.
Gross and colleagues were the first of many investigators to describe the use of aortopexy specifically for treating TBM caused by a vascular anomaly. In this procedure, the ascending aorta is approached, traditionally through a right-sided anterior thoracotomy at the third intercostal space, or through alternative routes . Traction on the sutures placed in the wall of the aorta juxtaposes it to the undersurface of the sternum, which also pulls the anterior wall of the trachea forward. This mechanical fixation of the aorta widens the anteroposterior dimensions of the trachea, and prevents collapse. Aortopexy is not always successful in relieving the collapse and therefore, additional surgical approaches have been devised.
External splinting with autologous materials, most commonly a resected rib, and prosthetic materials, such as silastic, membrane-reinforced, crystalline polypropylene and high-density polyethylene (Marlex, Phillips Petroleum, Bartlesville, Oklahoma) mesh, have been used to support the flaccid trachea . Support can be provided either by suturing a rigid support directly to the membranous trachea through a posterolateral right thoracotomy or by wrapping the supporting material around three quarters of the tracheal circumference through an anterior cervical or median sternotomy approach. Hagl and colleagues described bronchoscopically guided, external tracheobronchial suspension within a ring-reinforced polytetrafluoroethylene prosthesis, which immediately relieved severe malacia of the trachea or main bronchi in infants without necessitating resection. In animal studies and in limited human studies, this procedure did not affect the growth of the trachea adversely. However, it is an invasive procedure that may not treat distal bronchial lesions adequately and may not be well-tolerated by patients who have complicated conditions. Internal tracheal stent placement with a silicone prosthesis was first attempted in 1965 by Montgomery. Since then, various stents have been used, including those made of silicone and metal. The obvious advantages of stents are their less invasive nature and the shorter surgical recovery times. However, complications include granulation tissue formation, difficult removal, stent fracture, the need for additional stent placement, migration, the need to further dilate the stent as the child grows, and death. An advantage of the silicone stent is its relatively easy removal and the deployment of a larger stent, if required, as the child grows. Metallic, self-expanding stents have been catastrophic in many cases. Removability is especially important because children stand a reasonable chance of becoming asymptomatic once the airway grows. Currently, stents are used in limited situations in which conventional therapy has failed. However, stents currently in development, such as resorbable biopolymer stents , may address the limitations of those in current use.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


