Congenital Fundus Abnormalities
Eric Chen
Mitchell S. Fineman
Gary C. Brown
Because most of the congenital fundus abnormalities are caused by interruptions in the orderly development of the eye, a brief review of ocular embryology is necessary.
Embryology of the Eye
The earliest embryonic structure in which the three germ layers (endoderm, mesoderm, and ectoderm) are recognizable is known as the embryonic plate.1,2 Running longitudinally in the center of the embryonic plate is the neural groove, along each side of which develops an ectoderm-covered neural ridge. At the anterior end of each neural ridge a protuberance called an optic vesicle develops (Fig. 1). These spherical outpouchings can be seen at about the 4-mm (<4-week) gestational stage.
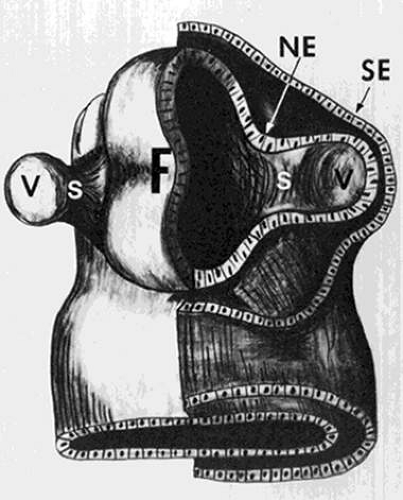 FIGURE 1. Schematic drawing of the forebrain (F) and optic vesicles at about the 4-mm gestational stage. A cross section of one vesicle shows neural ectoderm (NE) and surrounding surface ectoderm (SE). The vesicle (V) eventually develops into the globe, and the optic stalk (S) becomes the retrobulbar optic nerve. (Adapted from Mann I: Development of the Human Eye. New York: Grune & Stratton, 1969 with permission.) |
The optic vesicle is composed of neural ectoderm and is surrounded by surface ectoderm (Fig. 1). At about the 5-mm stage, both vesicles invaginate, and the outer wall of each becomes the inner wall of the newly formed concavity, the optic cup (Fig. 2). This inner wall of the optic cup (the former outer wall of the optic vesicle) gives rise to the retina, and the outer wall of the optic cup forms the retinal pigment epithelium (RPE).
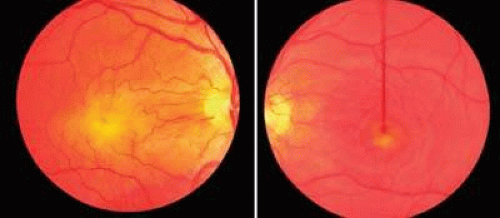 FIGURE 2. The optic cup, which is formed by an invagination of the outer wall of an optic vesicle. The vertical opening present in the inferior half of the optic cup is the embryonic fissure (EF). The optic stalk (S) is shown, and the fissure can be seen extending posteriorly along its inferior aspect. (Adapted from Mann I: Development of the Human Eye. New York: Grune & Stratton, 1969 with permission.) |
As the optic vesicles invaginate to form the optic cups, a groove remains open on the inferior aspect of each (Fig. 2). Known as the embryonic fissure, this opening is the pathway by which the mesodermal tissue, which gives rise to the intraocular vascular supply, enters the globe. Mesodermal tissue also surrounds the optic vesicles and eventually forms the choroid. Apparently, the choroid develops only in areas where the mesoderm is in contact with the RPE. This concept is supported by the clinical fact that in eyes with a retinochoroidal coloboma secondary to faulty closure of the embryonic fissure, the choroid, as well as the RPE, fails to develop properly. Closure of the embryonic fissure begins centrally and is normally complete at about the 13-mm stage (5 to 6 weeks).
The earliest recognizable structure associated with the optic disc is the primitive epithelial papilla. This is simply an accumulation of cells from the inner layer of the optic cup that surround the superior end of the embryonic fissure. At or about the 17-mm stage, nerve fibers grow from the retinal ganglion cells through the primitive epithelial papilla into the optic stalk, and the optic nerve is thus formed.
The first vascular supply to the globe enters through the embryonic fissure as the hyaloid artery at about the 5-mm stage. Derived from the carotid artery, this vessel forms a network (vasa hyaloidea propria) that vascularizes the primary vitreous. The vasa hyaloidea propria is maximally developed at about the 40-mm stage (third month of gestation) and regresses, until even the intraocular hyaloid artery itself is usually gone at about the eighth month of gestation. The portion of the hyaloid artery running within the optic nerve becomes the central retinal artery within the nerve.
Developmentally, the retinal vascular system does not appear to arise directly from the hyaloid artery. The most widely accepted concept has been summarized by Ashton, who states that at about 15 to 16 weeks of gestation (100-mm stage) mesenchymal cells appear on the optic disc adjacent to the hyaloid artery.3 Cords of these cells invade the superficial retina and differentiate into endothelial cells, which form capillaries. At this time it is not possible to make a distinction between arteries and veins. In turn, the capillaries undergo a process of remodeling and the retinal arteries and veins are formed.
Vascular Abnormalities
Prepapillary Vascular Loops
Prepapillary vascular loops are blood vessels that project from the optic disc into the vitreous cavity and then return to the disc to continue their natural course. First described by Liebreich in 1871,4 the loops have at least one ascending and one descending limb, and 85% to 95% are arterial in origin.5 Occasionally, an arterial loop projects from the disc and returns to the retina, while its venous counterpart may arise from the retina and exit into the disc. Very rarely a loop arises from the retina and returns to the retina; this particular anomaly has been termed a preretinal vascular loop.6
The vessels may appear as simple hairpin loops (180-degree turns), in a figure-of-eight, or in a corkscrew configuration. In about 30% of cases, the loop is surrounded by a white, glial-appearing sheath (Fig. 3).7 The average arterial loop extends approximately 1.5 mm into the vitreous cavity, probably within Cloquet’s canal; the largest reported loop reached about 7.9 mm in height.8 Venous prepapillary loops are usually less elevated.9 In contrast to persistent hyaloid arteries, prepapillary vascular loops have not been observed to extend as far anteriorly as the posterior lens capsule.
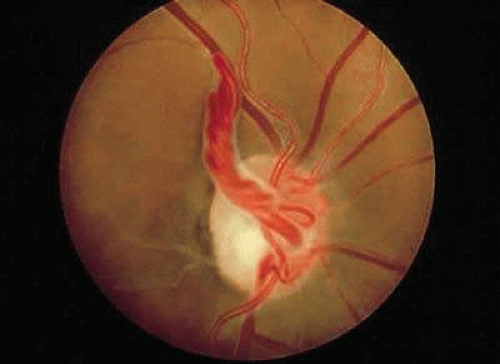 FIGURE 3. Prepapillary arterial loop in the left eye of a 30-year-old patient. The loop is encased by a white sheath that has a glial appearance. |
Embryologically, prepapillary loops are thought to occur at about the 100-mm stage, when the retinal vessels are developing. For some unknown reason, a vessel probably grows into Bergmeister’s papilla, which is maximally developed at about the 180-mm stage, and then returns to the retina. It has been proposed that the loop requires Bergmeister’s papilla as a scaffold for growth.5 Thus, its growth is limited, because Bergmeister’s papilla usually does not extend more than one third of the distance into the vitreous cavity.
Prepapillary loops are rare; the incidence ranges from 1 in 2,000 to 1 in 9,000 patients.10,11 Bilaterality occurs in about 9% to 17% of cases of arterial loops, but the percentage with venous loops is uncertain. Arterial prepapillary loops most commonly supply the inferior retinal vascular system, in contrast to venous prepapillary loops, which usually drain the superior retinal vascular system.12
Upon fluorescein angiography, prepapillary loops demonstrate rapid flow. However, there may be a sector delay in filling of the optic disc or the area of retina supplied by the loop due to the increased distance that blood must travel through the loop. Prepapillary loops are not typically seen in association with any other fundus anomalies except for cilioretinal arteries, which have been seen in up to 75% of eyes with prepapillary loops.11 There are singular reports of the simultaneous presence of prepapillary arterial and venous loops,13 change of regressed and patent segments of a retinal artery associated with a prepapillary arterial loop over time,14 and formation of a macroaneurysm on a prepapillary loop.15 Ocular complications associated with prepapillary vascular loops include: branch retinal artery obstruction in the area of retina supplied by the loop,11,16 amaurosis fugax, and subretinal, retinal, preretinal, or vitreous hemorrhage.11,17,18,19 Presumably, kinking of the loop and impairment of blood flow dynamics in some way contributes to retinal artery obstruction. Other reported occurrences of obstruction associated with prepapillary loops have implicated transient increases in intravenous pressure secondary to a Valsalva-like mechanism, which may further raise hydrostatic pressure in the loops20, and carotid artery stenosis, which reduces perfusion pressure across the loop and further increases the risk of loop thrombosis.21 Why hemorrhage occurs is uncertain, although we have noted it to occur in conjunction with acute posterior vitreous detachment. Fluorescein angiogram reveals no vessel leakage, and spontaneous resorption of the blood and an excellent prognosis is the rule.22 No consistently associated systemic abnormalities have been found in conjunction with prepapillary loops.
The differential diagnosis of arterial prepapillary loops includes persistent hyaloid artery. However, the latter is only a single vessel, without ascending and descending branches. Congenital venous prepapillary loops must be differentiated from the acquired variety.23 Congenital loops are usually single and unassociated with other ocular abnormalities, while acquired venous loops are often multiple and seen with disease entities such as retinal venous obstruction and optic nerve tumors.24
Persistent Hyaloid Artery
The hyaloid artery enters the globe through the embryonic fissure at about the 5-mm gestational stage. It branches throughout the vitreous cavity to form the vasa hyaloidea propria, the vascular supply of the primary vitreous. Additionally, it contributes to the tunica vasculosa lentis to supply blood to the developing lens.25 Maximally developed at about the 40-mm stage (9 weeks), the vasa hyaloidea propria begins to regress at the 60-mm stage (11 weeks). The hyaloid artery becomes impervious to blood at about 7 months, and the end of the involutional process usually occurs during the eighth month of fetal life, at which time the intravitreal artery becomes atrophic.26 The sequence of regression is such that the vasa hyaloidea propria regresses first, followed in turn by the capillaries of the tunica vasculosa lentis and then the hyaloid artery.27
The hyaloid artery can be seen in up to 90% of premature infants and 3% of infants born at term, and occasionally the vessel will persist into adult life.28 Ophthalmoscopically, posterior remnants of a persistent hyaloid artery can appear as a single vessel extending from the optic disc anteriorly through Cloquet’s canal or as elevated glial tissue at the optic disc (Bergmeister’s papilla). The hyaloid artery may be filled with blood, but is usually bloodless after birth and can extend as far anteriorly as the posterior capsule of the lens (Fig. 4). Usually the insertion on the posterior capsule is located inferonasal to the visual axis. Occasionally, after the vessel has regressed, only the circular point of insertion remains; this is known as a Mittendorf dot.
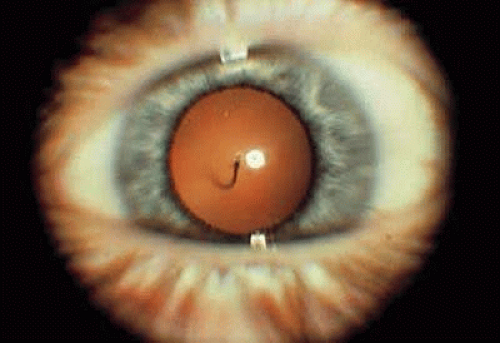 FIGURE 4. Persistent hyaloid artery inserting inferonasally on the posterior capsule of the lens in this left eye. |
A persistent hyaloid artery has been found in association with persistent hyperplastic primary vitreous and retinopathy of prematurity29,30,31,32 and should be considered a possible complicating factor during surgery.33 Indeed, intraoperative diathermy of a persistent hyaloid artery has been reported to result in a branch retinal artery occlusion.34 Vitreomacular traction and epiretinal membrane formation secondary to a persistent hyaloid artery has been described,35 and prepapillary and vitreous hemorrhage has been seen in association with persistent hyaloid arteries. Poor visual outcomes occur secondary to strabismus, nystagmus, and progression of Mittendorf’s dot to cataract. Possible mechanisms for hemorrhage include: rapid eye movements during sleep, trauma, or spontaneous bleeding.33,36,37,38,39,40
Bergmeister’s Papilla
Although technically not a vascular anomaly, Bergmeister’s papilla develops around the posterior aspect of the fetal hyaloid artery and appears as elevated glial tissue at the disc. Arising from the primitive epithelial papilla, neuroectodermal cells proliferate until they are maximally developed, at about the 180-mm stage (5 to 6 months).1 The glial sheath that forms around the proximal one third of the hyaloid artery begins to atrophy during the seventh month of intrauterine life, and the point to which it regresses determines the character of the physiologic cup. Failure of complete regression produces a persistent Bergmeister’s papilla, and may be associated with absence of the physiologic cup and with congenital anomalies such as prepapillary vascular loops. Visual acuity is not affected, and systemic associations are lacking.
Retinal Macrovessels
Retinal macrovessels are abnormally large single blood vessels, usually veins, which traverse the macular region and supply or drain the retina both inferior to and superior to the horizontal raphe (Fig. 5). This aspect of blood supply or drainage is most unusual, because only approximately 1% of eyes have a retinal vessel that extends to any degree on both sides of the horizontal raphe.41 Relatively large branches from these vessels may penetrate the foveal region, and small branches are occasionally seen traversing the foveal avascular zone. The aberrant retinal vessel is congenital and usually unilateral. Surprisingly, most eyes with this abnormality have normal visual acuity and are detected on routine examination.42,43
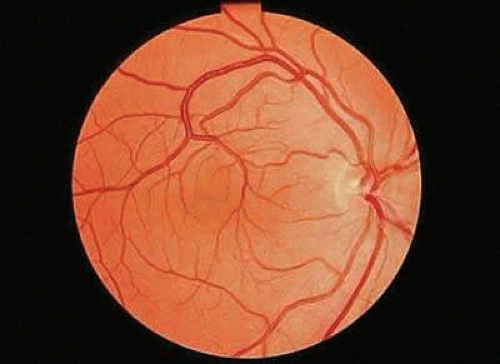 FIGURE 5. Venous congenital retinal macrovessel in the right eye. Abnormally large vessels are present in the foveal region, and the vein drains the retina on both sides of the horizontal raphe. |
Upon fluorescein angiography, small arteriovenous communications have been noted in some eyes with macrovessels.42,44 Archer and associates have referred to such eyes as having a group I, or mild, arteriovenous communication.44 Patients classified as group II have a more pronounced communication, with large vessels both entering and leaving the optic disc, and in group III the malformation is so pronounced that visual acuity is decreased as optic nerve tissue is replaced by enlarged vascular elements (Fig. 6). The abnormalities seen in groups II and III are usually referred to as racemose angiomas rather than macrovessels. Patients with anomalies characteristic of these latter two groups, in association with vascular malformations of the skull or central nervous system, are considered to have the Wyburn-Mason syndrome.45 However, eyes with congenital retinal macrovessels do not necessarily have arteriovenous communications, and systemic abnormalities have not been associated with these milder vascular abnormalities. Although the previously described arteriovenous anomalies are thought to be congenital, remodeling of some of the more bizarre malformations (group III) has been observed.46
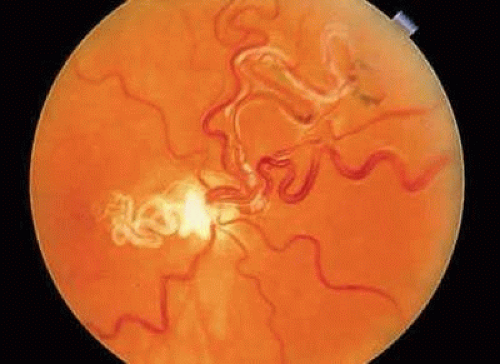 FIGURE 6. Right eye of a 20-year-old patient with a large congenital arteriovenous anastomosis, also referred to as a racemose angioma. The visual acuity in this eye was no light perception, and additionally the patient had an arteriovenous malformation in the jaw, which produced severe bleeding at the time of dental extraction. (Courtesy of Dr. Jerry Shields.) |
Fluorescein angiography of congenital retinal macrovessels (group I) commonly demonstrates small areas of retinal capillary nonperfusion.42 These may be located in the foveal region or in a perivascular distribution surrounding the abnormal vessel. Flow through the vessels appears to be rapid, although delayed emptying of dye may be observed.42,43 Permeability alterations of the anomalous vessel is not a feature of this disorder.47
Typically, visual acuity is unaffected, although decreased visual acuity with congenital macrovessels may be secondary to the mere presence of the macrovessel in the foveal area, foveal cysts, or preretinal hemorrhage. Hemorrhage in the setting of Valsalva retinopathy48 and repetitive rollercoaster rides, which may cause venous decompensation secondary to dramatic fluctuations in venous pressure,49 has been reported. Also, in one report a patient developed a serous retinal detachment secondary to vitreous traction on an arteriovenous anastomosis, with spontaneous resolution following posterior hyaloid detachment.50 Vision and fundus findings appear to remain stable over time. Other ocular abnormalities include: a poor foveal reflex, minor displacement of the fovea, and retinal pigment epithelial mottling.42,51,52 Gass described one patient with congenital retinal macroveins who had similar vascular anomalies of the conjunctiva and tongue,47 and a congenital macrovessel has been reported in association with a congenital hamartoma of the retinal pigment epithelium.53
Cilioretinal Artery
Cilioretinal arteries are vessels that supply the retina but are derived from the short posterior ciliary arteries and, rarely, directly from the choroidal vessels, rather than from the central retinal artery.54 Anastomoses between cilioretinal- and central-retinal-derived circulations are minimal. Typically, cilioretinal arteries appear to emerge from the optic disc or disc margin separately from the central retinal artery. A characteristic hook, which has been likened to a walking stick handle, is another helpful ophthalmoscopic sign (Fig. 7).55
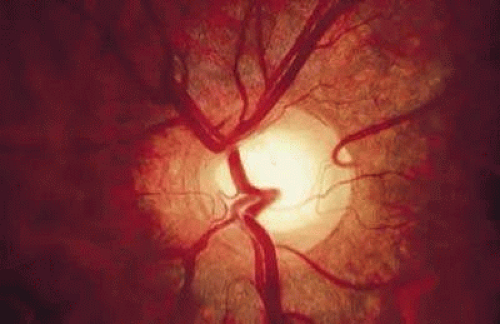 FIGURE 7. Cilioretinal artery emerging from the optic disc at the 2 o’clock position. Note the characteristic hook made by the vessel at its origin on the disc margin. |
Cilioretinal arteries are considered to be the most common retinal vascular anomaly, and their presence in healthy persons may be influenced by genetic factors.56 With ophthalmoscopy alone, cilioretinal arteries have been noted in about 20% of eyes.57,58,59 Fluorescein angiography has demonstrated that cilioretinal arteries are present in about 32% of eyes.60 Usually a cilioretinal artery will fill with the choroidal circulation 1 to 2 seconds prior to the appearance of dye within the retinal arterial system.
In healthy eyes, the presence or absence of a cilioretinal artery is clinically insignificant, as cilioretinal arteries usually supply only a small portion of the fundus and are most often confined to the macula. In about 1 eye in 2,900, a cilioretinal artery may supply a major part of the fundus.61 However, when prepapillary arterial loops are present, prominent cilioretinal circulations are commonly seen.11,62 Cilioretinal arteries supply the foveal region in about 10% of eyes.63 This is clinically relevant in that such a cilioretinal vessel may preserve central vision in eyes that sustain a central retinal artery obstruction.63 A cilioretinal artery has also been reported to be the main feeder vessel in a case of retinal angiomatous proliferation.64 The presence of a temporal cilioretinal artery has also been correlated with increased retention of central visual field and visual acuity in eyes with advanced open-angle glaucoma, possibly because of an improved blood supply to the prelaminar portion of the optic disc.65 Conversely, cilioretinal arteries may also become obstructed and produce a central field defect.66 Cilioretinal artery obstructions account for approximately 5% of arterial obstructions in the retina.47,55 In general, isolated cilioretinal artery obstruction (44%) carries a favorable visual prognosis with 90% of eyes achieving a visual acuity of 20/40 or better.67
Cilioretinal artery obstruction may also occur in association with central retinal venous obstruction (44%) or ischemic optic neuropathy (12%).67,68 Central retinal vein occlusion is thought to raise intraluminal resistance in the cilioretinal artery and subsequently cause occlusion.69,70 Other investigators have documented abnormal central retinal artery inflow by intravenous fluorescein angiography in patients with this disorder.71 These eyes have a less favorable prognosis with 70% of eyes achieving a visual acuity of 20/40 or better.67 A cilioretinal artery obstruction occurring in the setting of ischemic optic neuropathy may be explained by the shared origin of their vascular supply.68 These eyes have the worst prognosis with no eyes achieving better than 20/40 visual acuity.67 Cilioretinal artery obstruction has also been reported in association with posterior scleritis,72 following carotid artery dissection,73 and following procedures including LASIK,74 cardiac catheterization75, and embolization of an intracranial meningioma.76
Optic Disc Abnormalities
Excavated Optic Disc Anomalies
Congenital Optic Pits
Congenital pits of the optic nerve head are rare, occurring in about 1 in 11,000 patients.77 They appear as round or oval localized depressions within the optic disc (Fig. 8), typically do not reduce visual acuity, and may be associated with reduced retinal fiber layer thickness at the corresponding papillomacular bundle.78 Over one half are positioned temporally on the nerve head, while about one-third are located more centrally on the disc.5 In 95% of eyes with noncentrally located pits, peripapillary chorioretinal disturbances are seen adjacent to the anomaly.79 Optic pits range in size from 0.1 to 0.7 disc diameters along their widest dimensions and may be as deep as 25 D, although the mean depth is about 5 D.79 Bilateral involvement is seen in about 15% of patients. In those with unilateral pits, the optic disc itself is larger in the affected eye about 80% of the time.79
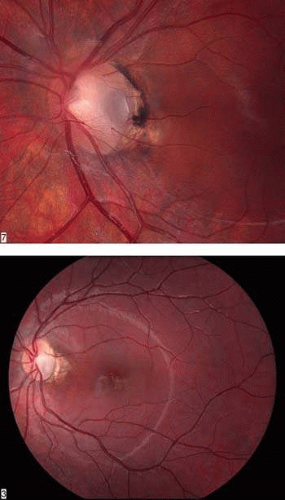 FIGURE 8. Congenital pit of the optic nerve head. A. The pit is gray and inferotemporally located, and it has what appears to be a cilioretinal artery emerging from its lateral border. Mild peripapillary chorioretinal changes are evident adjacent to the pit. B. Macular retinal detachment secondary to a congenital optic nerve pit. |
Embryologically, congenital optic pits probably arise from defective formation of the primitive epithelial papilla. However, in some cases a pit is located inferiorly and is associated with a retinochoroidal coloboma. In these latter instances, it may arise from defective closure of the superior aspect of the embryonic fissure.80 Histologically, optic pits contain herniations of dysplastic retina into a collagen-lined pocket through a defect in the lamina cribrosa.5
Petersen was probably the first to realize that the association between congenital optic pits and serous retinal detachment was more than coincidental.81 Other congenital cavitary anomalies of the optic nerve, such as optic nerve coloboma and morning glory disc anomaly, can also be associated with serous macular detachments and may lie on a spectrum of disease. The serous retinal detachment is usually confined to the macular region and rarely exceeds 1.5 mm in height. Approximately 40% of pits reported have been noted to have such a nonrhegmatogenous detachment.5 The great majority of detachments are seen in eyes with larger or temporally located pits, and the mean age at onset is about 30 years.79 The initial step appears to be accumulation of intraretinal fluid emanating from the disc anomaly, leading to a retinoschisis-like separation with cystic changes in the retinal stroma, but without the expected complete loss of retinal function in the affected area.82,83,84. Approximately one third of these eyes can have subretinal precipitates on the outer surface of the detached retina. This schisis-like separation precedes macular detachment and always communicates with the optic disc, even if the serous detachment does not. Optical coherence tomography suggests that breakthrough of this fluid into the subretinal space through a lamellar, outer layer macular hole or minute invisible breaks in the outer retina then produces macular detachment.79,85,86,87 Vitreoretinal traction may contribute to macular detachments, because in one case a macular break developed after minor head trauma in a patient with an optic nerve pit and serous macular detachment,88 and alleviating vitreoretinal traction by pars plana vitrectomy or posterior scleral buckling with or without laser retinopexy has resulted in resolution of the subretinal fluid.89,90,91,92
Controversy exists as to the etiology of the subretinal fluid. Possible sources include: (a) vitreous fluid by way of the pit or a macular hole, (b) cerebrospinal fluid, (c) leakage from vessels within the pit, and (d) leakage from choroidal vessels. In collie dogs, a direct communication has been demonstrated between the vitreous cavity and the subretinal space by way of the pit.93 In humans, intraoperative drainage of subretinal fluid through the pit94,95 and subretinal migration of vitreous substitutes such as gas or oil through cavitary disc lesions96,97 offer clinical confirmation of a similar pathway, although histopathologic correlation is lacking. Some optical coherence tomography studies85,86 and clinical findings in patients with optic pits (intracranial migration of silicone oil)98 and morning glory anomaly99,100 suggest that the subretinal fluid may be derived from cerebrospinal fluid from the perineural subarachnoid space. A possible unifying theory accounts for variable connections between the vitreous, subarachnoid, and subretinal spaces and transmission of intracranial pressure differences to the pit via the subarachnoid space.94 Vascular sources for macular detachments are less likely. Although fluorescein angiography reveals leakage originating from vessels within the pit more commonly in eyes with a serous retinal detachment,101 many eyes with leakage do not have subretinal fluid. The presence of cilioretinal arteries, which are present in 60% of eyes with optic nerve pits, are also associated with leakage of intravenous fluorescein dye.79,102
Fluorescein angiography usually demonstrates early hypofluorescence of a pit, and 50% progress to late hyperfluorescence; this is strongly correlated with the presence of cilioretinal arteries arising from the pit102 (Fig. 9). Visual fields are abnormal in about 60% of eyes with pits; enlarged blind spots, arcuate scotomas, and altitudinal defects are most commonly encountered.79
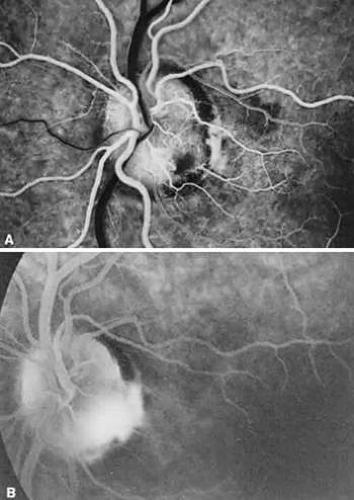 FIGURE 9. A. Fluorescein angiogram in the laminar venous filling phase of an eye with an inferotemporal optic pit. The pit is hypofluorescent at this time. B. Ten minutes after the injection, the pit is hyperfluorescent. |
Although spontaneous resolution can occur in around 25% of untreated patients, without treatment, the visual prognosis is poor in most patients with an optic nerve pit and serous detachment of the macula.79,103,104 One study found that over 50% of eyes with retinal detachment and a minimum follow-up of 1 year have an acuity of 20/100 (6/30)* or worse.79 These findings were confirmed by another study where 80% of eyes with an optic nerve pit and serous macular detachment had a visual acuity of 20/200 or worse after an average follow-up period of 9 years.104 Visual loss occurs within 6 months of the development of the serous macular detachment and is due to cystic retinal degeneration, macular hole formation, and retinal pigment epithelium atrophy.
Laser photocoagulation to the peripapillary retina adjacent to the pit has been used for the treatment of associated macular retinal detachment with variable success.105,106,107,108 Although this therapy sometimes promotes resolution of the subretinal fluid, it is uncertain whether the visual acuity improves compared with the natural course of the disease.5 Pars plana vitrectomy and intraocular gas tamponade with or without laser treatment are successful in reattaching the macula and improving central vision in most macular detachments secondary to optic nerve pits.89,109,110,111,112,113,114,115,116 Other reported methods include macular buckling, which has demonstrated postoperative improvements in visual field and multifocal electroretinogram studies,117,118 and a case report of inner retinal fenestration.119
No consistent systemic abnormalities have been associated with pits, but the occurrence of a basal encephalocele and agenesis of the corpus callosum in a patient with a pit has been noted120,121 Although no definite inheritance pattern has been identified for pits, familial cases, autosomal dominant cases, and cases occurring in monozygotic twins have been seen.122,123,124
Optic Nerve Hypoplasia and Aplasia
Optic nerve hypoplasia is the most common congenital disc anomaly, occuring in 6.3 out of 100,000 children, and is the leading single cause of blindness in infants and toddlers.125 There is probably a spectrum of hypoplasia ranging from mild hypoplasia of the optic nerve head to absence of the nerve and retinal vessels (aplasia). Typically, a hypoplastic optic disc appears small and pale and is partially or totally surrounded by a yellow white ring that may be variably pigmented (Fig. 10). Because of the encircling ring, these eyes have been referred to as having the double-ring sign.126 The outer border of the yellow-white ring denotes the juncture of the sclera with the lamina cribrosa and typically corresponds to the size of a normal optic nerve head. The retinal vessels are usually of normal caliber. Histopathologic examination shows a diminished nerve-fiber layer in nearly all eyes and usually a reduced number of ganglion cells.126,127 The outer retinal layers appear normal. An unusual variant of optic nerve hypoplasia occurs in children with periventricular leukomalacia, who have a normal-sized optic disc with a large cup and no associated endocrine dysfunction.128
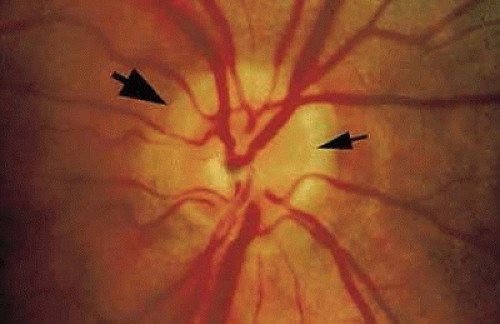 FIGURE 10. Hypoplastic optic disc. The outer ring (large arrow) represents the junction of the sclera and lamina cribrosa, and the inner ring (small arrow) corresponds to the termination of the retina and retinal pigment epithelium, both of which abnormally extend centrally over the lamina cribrosa. |
In recent reports, bilateral involvement far exceeds unilateral optic nerve hypoplasia. In bilateral cases there is no apparent sex predilection,129,130,131,132 but over three-fourths of patients are men in unilateral cases.133 Visual acuity in involved eyes may range from normal to no light perception, although most cases are in the counting-fingers to light-perception range,129,130,133 and localized visual field defects are often combined with generalized constriction. Despite this fact, it has been suggested that a trial of occlusion therapy is warranted in cases of unilateral hypoplasia associated with strabismus.133 Strabismus is often seen in unilateral cases, usually in the form of an esotropic deviation.129,130,134 It may also be found in bilateral cases, but in these eyes a pendular nystagmus is more frequently encountered, due to poor visual acuity.129,130 Careful retinoscopy and refraction is essential in these patients, because high rates of astigmatism have been found in affected eyes.135,136 Other ocular findings include increased vascular tortuosity and a low number of vascular branching points132, and other reported ocular associations include aniridia,137 optic nerve diastasis,138 microphthalmos, and ptosis. One report suggests isolated retinal vein tortuosity may be a marker for endocrinopathy.139 Electroretinography is usually normal in eyes with this disorder.140
Calculation of the disc-macula: disc diameter ratio has been recommended as a means of establishing the diagnosis of optic nerve hypoplasia.141 A ratio of 3.0 or higher has been found to be correlated with the diagnosis of optic nerve hypoplasia, although this may not be as accurate in the neonatal period or in cases of segmental optic nerve hypoplasia.142,143,144 Magnetic resonance imaging and B-scan ultrasonography can also demonstrate a small optic nerve in affected individuals.145,146 Systemic abnormalities are more commonly associated with bilateral optic nerve hypoplasia and may include anencephaly, hydranencephaly, chromosomal abnormalities (trisomy 18 and 5p deletion), and midline cerebral defects such as septo-optic dysplasia (De Morsier syndrome), as well as others.126,127,130,147,148,149,150 Septo-optic dysplasia refers to the association of optic nerve hypoplasia, absence of the septum pellucidum,151 and thinning or agenesis of the corpus callosum. Pituitary dwarfism152 and diabetes insipidus153 can also be seen with the syndrome. At least one quarter of patients with bilateral optic nerve hypoplasia have hypothalamic-pituitary endocrine dysfunction and 11.5% have De Morsier syndrome.154,155 Pituitary hormone insufficiency is important to recognize, as untreated, it can lead to hypoglycemia, growth retardation, and even sudden death.156 Magnetic resonance imaging of affected individuals with unilateral or bilateral optic nerve hypoplasia can provide prognostic information regarding the likelihood of neurodevelopmental deficits and hypothalamic-pituitary dysfunction as well as detect other intracranial abnormalities.136,157,158,159,160
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


