Congenital Craniofacial Anomalies and Their Management
Brian J. Forbes
James A. Katowitz
William R. Katowitz
Congenital abnormalities of the cranium and face present complex diagnostic and therapeutic challenges to the ophthalmologist. Patients with craniofacial anomalies are best treated by a multidisciplinary team that includes specialists from plastic surgery, neurosurgery, ophthalmology, otolaryngology, oral surgery, orthodontics, anesthesia, and genetics as well as specialists in the disciplines of psychiatry, social work, and nutrition. These specialists must work together to provide for the overall well-being of the patient. To assist in the evaluation and treatment of these patients, the ophthalmologist must possess an understanding of craniofacial syndromes as well as the necessary medical and surgical interventions required to improve ocular and adenexal problems. This chapter systematically reviews the major craniofacial anomalies of ophthalmic importance and highlights salient treatment issues. Craniofacial syndromes may be conveniently divided into two broad categories: craniosynostosis or premature closure of the cranial sutures and clefting syndromes.
To facilitate understanding of the congenital craniofacial anomalies, a review of the normal embryogenesis of the human head is valuable. Although a more complex review of embryology is beyond the scope of this chapter, essential morphogenetic events are presented here to help the reader better visualize the formation of the craniofacial anomalies.
Embryology
The fourth to eighth weeks of gestation are referred to as the embryonic period. The face develops mainly between the fifth and eighth weeks and, by the end of the eighth week, the embryo has a human appearance. Early in the fourth week, the embryo is a straight flat structure with a dorsal neural tube (Fig. 41.1). Formation of the neural tube begins in the middle of the embryo and progresses toward the cranial and caudal ends. As the neural tube separates to a more ventral position from the surface ectoderm, a subset of neuroectodermal cells migrate to the dorsolateral aspects of the tube to become the neural crest. Neural crest cells ultimately give rise to the connective tissue elements of the eye and adnexa, including corneal keratocytes, iris stroma, corneal stroma, ciliary smooth muscle, sclera fibroblasts, optic nerve meanings, orbital fibroadipose tissue, orbital nerve Schwann cells, and orbital bone. In addition, early in the fourth week of gestation, the paraxial mesoderm forms into longitudinal columns of paired cuboidal bodies called somites that laterally flank the neural tube. These somites are destined to form most of the paraxial skeleton and associated musculature.
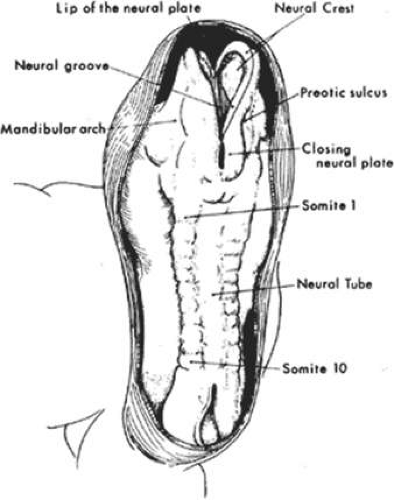 Figure 41.1. Early fourth-week embryo demonstrating cephalad open neural groove, closing neural plate, and flattened structure. (From Fries PD, Katowitz JA: Congenital craniofacial anomalies of ophthalmic importance. Surv Ophthalmol 35:87, 1990, with permission from Elsevier Science.) |
As the rostral neural groove closes, the first branchial arch begins to appear along the lateral portion of the neural tube. The first branchial arch, or mandibular arch, forms two paired mandibular processes, which will eventually fuse to become the mandible. At the superior lateral portion of each mandibular process, a small bud develops into the maxillary process, which eventually forms the maxilla, zygomatic bone, and squamous part of the temporal bone (Fig. 41.2). At the midfourth week, after neural tube invagination, the primitive cavity or stomodeum is flanked by five distinct facial processes: two paired mandibular processes, two paired maxillary processes, and a central frontonasal process. Most congenital malformations of the head and neck originate as the branchial arches transform into their adult derivatives.
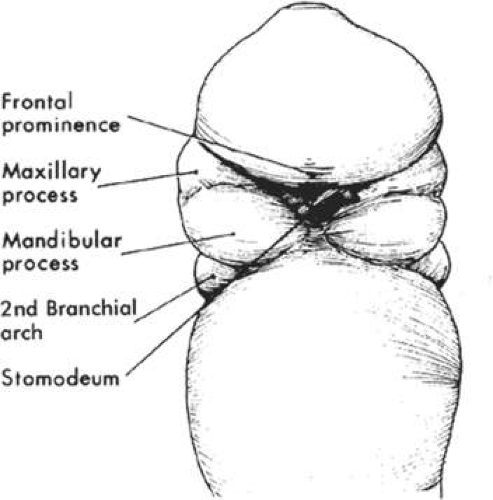 Figure 41.2. Midfourth week embryo after neural tube invagination. Maxillary, mandibular, and frontocranial processes are beginning growth into intended facial position. (From Fries PD, Katowitz JA: Congenital craniofacial anomalies of ophthalmic importance. Surv Ophthalmol 35:87, 1990, with permission from Elsevier Science.) |
The branchial arches are initially composed of an external layer of ectoderm and an internal core of endoderm. The migration of neural crest cells into the branchial arches causes them to enlarge. The ectodermal furrows dividing the facial processes are eventually obliterated by proliferating neural crest cells and mesenchyme. Fusional failure of the facial processes caused by the absence of neural crest cell migration is one of the proposed causes of “true” facial clefts and is described in more detail in later sections. At the beginning of the fourth week, the developing eye appears as a placode on the lateral forebrain and by the end of the fourth week there is an easily identifiable optic vesicle.
During the fifth week of gestation, the maxillary processes continue to enlarge and laterally border the recently perforated stomodeum (Fig. 41.3). The nasal placodes, which appear late in the fourth week, transform into olfactory grooves. Horseshoe-shaped medial and lateral nasal prominences develop and the nonfused inferior portion of the olfactory groove remains continuous with the stomodeum. Also during the fifth week, six swellings, called auricular hillocks, develop from the first and second branchial arches and represent the rudimentary external ear. As the optic vesicle continues to grow, the overlying surface ectoderm differentiates into the lens placode. The optic vesicle invaginates to form the double-layered optic cup and the overlying surface ectoderm, with mesenchymal contributions from the neural crest cells, which will later form the eyelids (Fig. 41.4). The eyelids fuse at the ninth week and at the 25th to 26th weeks, the fused margins separate.
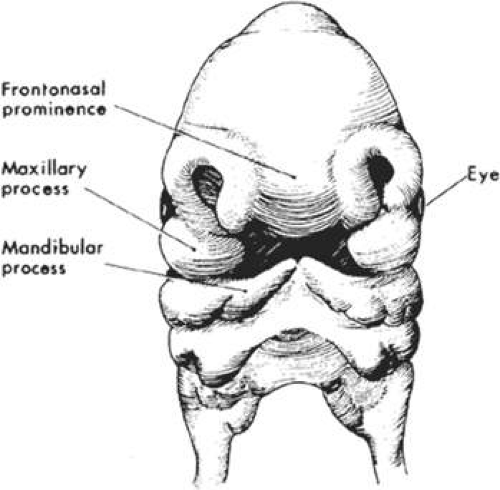 Figure 41.3. Frontal view, late fifth week embryo demonstrating prominent anteriorly located nasal pits with the nonfused inferior portion connecting to the recently perforated stomodeum. (From Fries PD, Katowitz JA: Congenital craniofacial anomalies of ophthalmic importance. Surv Ophthalmol 35:87, 1990, with permission from Elsevier Science.) |
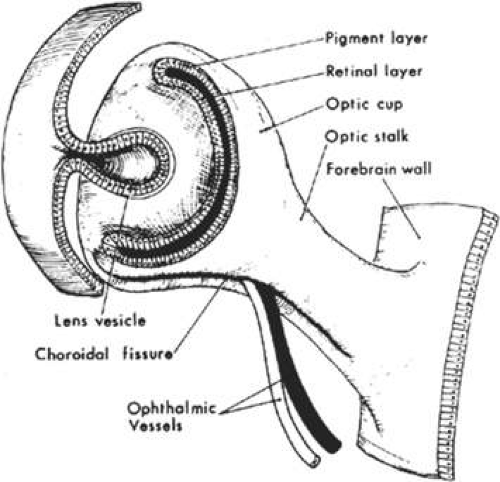 Figure 41.4. Late fifth week demonstration of ocular development showing lens vesicle, two-walled optic cup, and optic stalk with choroidal fissure incorporating the ophthalmic vessels. (From Fries PD, Katowitz JA: Congenital craniofacial anomalies of ophthalmic importance. Surv Ophthalmol 35:87, 1990, with permission from Elsevier Science.) |
In the sixth week, the medial nasal prominences fuse with each other and with the maxillary prominence (Fig. 41.5). Failure of the medial nasal prominence to merge with the maxillary prominence results in a cleft lip, with or without cleft palate, and the cleft lip may be unilateral or bilateral. Continued growth of the maxillary processes promotes the formation of the nasolacrimal groove along the lateral border of the nasal processes. The nasolacrimal groove will incorporate neuroectodermal cells, which will eventually become the nasolacrimal drainage apparatus. The mandibular process continues its growth and fuses at the midline by the end of the fifth week.
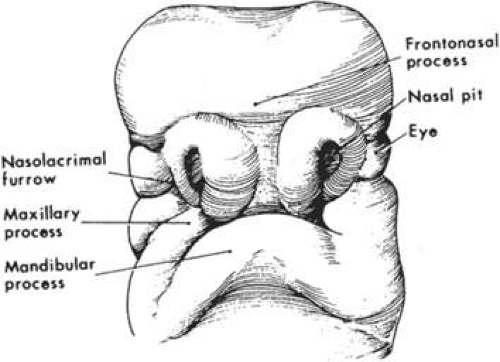 Figure 41.5. Sixth week embryo shows fusion of the maxillary process to the lateral nasal groove forming the nasal pit and nasolacrimal furrow. Note lateral position of developing eye. (From Fries PD, Katowitz JA: Congenital craniofacial anomalies of ophthalmic importance. Surv Ophthalmol 35:87, 1990, with permission from Elsevier Science.) |
During the seventh week of gestation, the maxillary processes fuse at midline, separating the nares from the mouth (Fig. 41.6). The enlargement of the maxillary process and expansion of the lateral head, combined with the horizontal collapse and the vertical elongation of the frontonasal process, allow for anterior rotation of the developing eyes. This anterior rotation of the eyes is not fully achieved until approximately 4 months of gestation and abnormalities in this process may result in hypertelorism (Fig. 41.7). By the seventh week, nearly all lower facial fissures and furrows have completely closed.
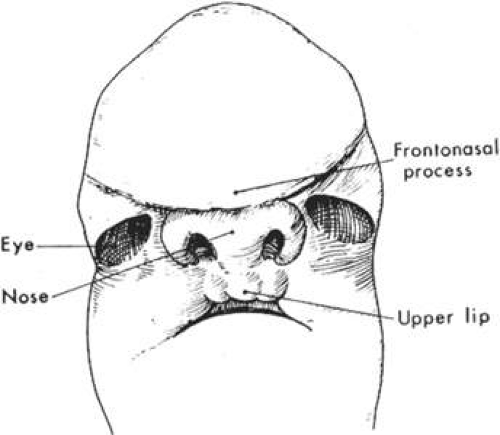 Figure 41.6. Seventh week embryo with completed maxillar and mandibular midline fusion, continued frontal rotation of the orbits and prominent frontonasal overhang. (From Fries PD, Katowitz JA: Congenital craniofacial anomalies of ophthalmic importance. Surv Ophthalmol 35:87, 1990, with permission from Elsevier Science.) |
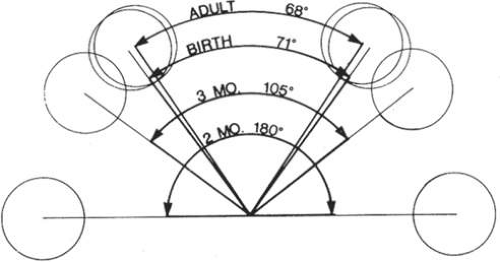 Figure 41.7. Separation of globes at 2 months’ gestation, 3 months’ gestation, birth, and adulthood. (From Fries PD, Katowitz JA: Congenital craniofacial anomalies of ophthalmic importance. Surv Ophthalmol 35:87, 1990, with permission from Elsevier Science.) |
By the eighth week of gestation, the embryonic face has distinct human characteristics despite the obvious differences in facial proportions. By the end of the embryonic period, external ears begin to assume their final shape despite being set low on the head. The eyes assume a more almond shape and continue their anterior rotation. The cranium loses most of its overhang while the chin and nose achieve a more pronounced profile.
In addition to an appreciation of the development of facial features, a fundamental understanding of the development of the bony skull is critical to an understanding of congenital craniofacial anomalies. The calvaria and cranial base represent the two major portions of the developing skull and have distinct embryonic causes. The calvaria arise from neural crest cell migration into cartilaginous condensations at the site of the future crista galli, lesser wings of the sphenoid, and petrous ridges. Primitive dura matter arises from these sites and is first observed at 5 to 6 weeks’ gestation. The dural sheaths expand to accommodate the rapidly enlarging brain and the junctions at which these dural sheaths appose determine the sites of the cranial sutures (Fig. 41.8). Intramembranous ossification of mesenchyme forms the bones of the cranial vault. Portions of the dura prevent closure of the cranial sutures and allow for expansion of the intracranial contents during fetal and infant growth. Premature fusion of one or more of the cranial sutures, termed craniosynostosis, produces various abnormal head shapes, which are discussed in more detail in later sections.
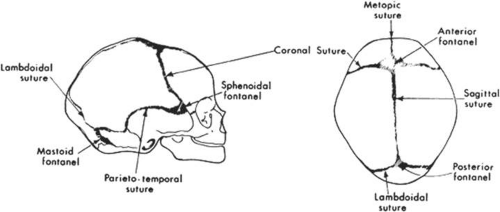 Figure 41.8. Normal sutures and fontanels of fetal skull. (From Fries PD, Katowitz JA: Congenital craniofacial anomalies of ophthalmic importance. Surv Ophthalmol 35:87, 1990, with permission from Elsevier Science.) |
At 4 to 5 weeks’ gestation, the neural crest cells responsible for formation of the cranial base first flank and later surround the notochord (Fig. 41.9). These include the alisphenoid, orbitosphenoid, and prechordal cartilage, which develop into the sphenoid, zygoma, temporal, and ethmoid bones. Premature fusion of these sutures, especially the sphenozygomatic and sphenoethmoidal sutures, can lead to shallow orbits and poor growth of the midface. The prechordal cartilage becomes the nasal septum, which, with ethmoid development, plays a vital role in the collapse of the wide embryonic interpalpebral distance. Variations in development of the ethmoid sinus and nasal septum may lead to hypertelorism.
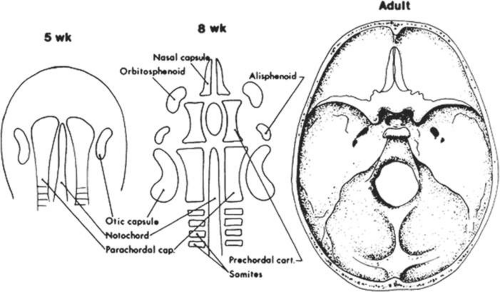 Figure 41.9. Schematic representation of cranial base cartilage and growth at weeks 5 and 8 of gestation. The alisphenoid, orbitosphenoid, and prechordal cartilages develop into the sphenoid, zygoma, temporal, and ethmoid bones in the adult skull. (From Fries PD, Katowitz JA: Congenital craniofacial anomalies of ophthalmic importance. Surv Ophthalmol 35:87, 1990, with permission from Elsevier Science.) |
Most congenital craniofacial anomalies arise from developmental abnormalities before the eighth week of gestation. Striking similarities between characteristics of the developing embryonic face and certain craniofacial deformities should now be readily apparent. It is as though growth and differentiation progress normally to a certain stage and then are abruptly interrupted. The following sections review in detail the craniofacial deformities most frequently encountered by the ophthalmology consultant.
Craniosynostosis
Craniosynstosis comprises a major group of congenital malformations in which one or more of the cranial sutures are closed prematurely. In 1851, the German pathologist Virchow first described how premature closure of one cranial suture promotes growth parallel to that suture and inhibits growth perpendicular to it.1 Although the terms craniosynostosis and craniostenosis are often used interchangeably, the preferred, more accurate term is craniosynostosis. The word craniosynostosis describes the process of premature sutural fusion, craniostenosis is the result.2
There are several different types of craniosynostosis. First, craniosynostosis can be defined as either simple or compound. Simple craniosynostosis refers to the premature closure of a single suture, whereas a compound craniosynostosis involves two or more sutures. The most prevalent type of craniosynostosis is scaphocephaly, in which the sagittal suture is synostosed and results in a boat-shaped skull. Boys are affected with craniosynostosis much more commonly than girls, whereas sagittal synostosis occurs in girls more often than boys.2
Craniosynostosis is also designated as either primary or secondary. In primary synostosis, the most common type, the cranial sutures are fused prematurely because of a genetic predisposition. In secondary craniosynostosis, the premature sutural fusion is secondary to another known disorder. Examples of secondary craniosynostosis include hematologic disorders (thalassemia and sickle cell disease), metabolic disorders (hyperthyroidism), metabolic disorders (mucopolysaccharoidosis), and malformations (microcephaly).2
Finally, craniosynostosis can be either isolated or syndromic. In the isolated form, the patient has no other abnormalities except those directly related to early sutural obliteration. In contrast, patients with the syndromic craniosynostosis have other primary morphogenic alterations. For example, in Apert syndrome, syndactyly of the hands and feet frequently occurs.
Specific terms, such as scaphocephaly, have been used to describe the typical skull deformities associated with various synostoses (Fig. 41.10). Single coronal sagittal, metopic, or lambdoidal synostosis, respectively, leads to various degrees of brachycephaly, scaphocephaly, trigonocephaly, or plagiocephaly. Oxycephaly (tower head) is used to describe multiple synostoses involving at least the coronal and sagittal sutures, with compensatory upward growth of the cranium. Triphyllocephaly3 or kleeblattschädel describes a cloverleaf-shaped skull, which is clinically characterized by the synostosis of the lambdoid and coronal suture with herniation of the cerebrum through a patent sagittal suture. The general term plagiocephaly refers to closure of a single cranial suture, whether sagittal, coronal, or lambdoidal and can lead to wide variability in cranial morphology. Although descriptive, these terms may be confusing and it is probably simpler to classify these patients by the sutures involved.4
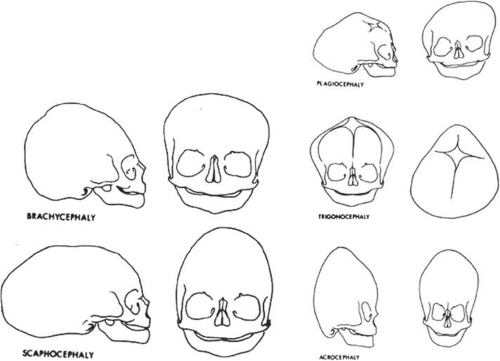 Figure 41.10. Frontal and lateral views of fetal skulls demonstrating more common calvarial abnormalities resulting from suture closure. (From Fries PD, Katowitz JA: Congenital craniofacial anomalies of ophthalmic importance. Surv Ophthalmol 35:87, 1990, with permission from Elsevier Science.) |
Traditional classification of craniosynostosis has been divided into three main groups: (a) craniofacial dystosis or Crouzon syndrome, (b) acrocephalosyndactyly or “Apert syndrome,” and (c) simple or single craniosynostosis. This classification system has largely been replaced by Cohen’s5,6,7 more extensive classification.8 Cohen’s classification of craniosynostosis categorizes these anomalies based on clinical similarities and genetic transmission5,6,7 (Table 41.1).
Table 41.1. Craniosynostosis Syndromes | ||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||
Three classic theories of pathogenesis explain craniosynostosis. Virchow1 believed that premature fusion of the cranial sutures was the primary dysgenesis and that the cranial base abnormalities were secondary to the craniosynostosis. Moss,8 conversely, postulated that the cranial base malformation was the primary event, resulting in secondary obliteration of the cranial sutures. Moss speculated that spatially malformed lesser sphenoidal wings in coronal synostosis, and a malformed cribriform plate in sagittal synostosis transmit aberrant tensile forces at points of dural attachments leading to premature fusion of the overlying sutures. Park and Powers9 implicated a primary defect in the mesenchymal blastema that produced both an abnormal cranial base and craniosynostosis.
There are chromosomal and environmentally induced causes for the craniosynostosis syndromes. The specific inheritance patterns and genetic anomalies of the individual craniosynostosis syndromes of ophthalmic importance are individually discussed in later sections. For many dysmorphic syndromes, it is possible to find families that display a particular phenotype in either an autosomal-dominant, recessive, or X-linked manner.10 In addition, patients with the same genetic condition may have fusion of completely different sutures.11 Nonsyndromic synostosis can be sporadic or familial, with similar suture involvement in several members of the same family.12 With the recent advancements in gene mapping and cloning techniques, investigation into the genetic elements of craniofacial dysmorphologies has advanced rapidly. It is critical for geneticists to be intimately involved with other craniofacial medical specialists so that they may provide genetic counseling to the patient’s family as well as further research, which may ultimately lead to possible genetic therapy.
Crouzon Syndrome
Crouzon syndrome, first described in 1912, is characterized by the triad of craniosynostosis, midface retrusion, and proptosis (Fig. 41.11).13 Crouzon syndrome, originally termed hereditarily craniofacial dysostosis, has a birth prevalence of 16.5 per million and accounts for 4.8% of all cases of craniosynostosis, making it the most common of the craniosynostosis syndromes.14 Crouzon syndrome has an autosomal-dominant inheritance pattern, with variable expression, and one-fourth of reported cases have a negative family history and thus presumably represent fresh mutations. Progress in our understanding of the molecular basis of the craniosynostoses accelerated after Crouzon syndrome was found to be tightly linked to markers on chromosome 10q25–q26.15,16 Evaluation of candidate genes in this region of chromosome 10 showed that mutations in the FGFR2 gene cause Crouzon syndrome as well as the FGFR3 gene when associated with acanthosis nigritans.17,18,19,20,21 The four known FGFRs make up a family of related, but distinct, tyrosine kinase receptors. They have similar protein structure, with three immunoglobulinlike domains in the extracellular region, a single membrane-spanning segment, and a cytoplasmic tyrosine kinase domain.22 Crouzon syndrome is seen in many different ethnic groups and is associated with advanced paternal age.16
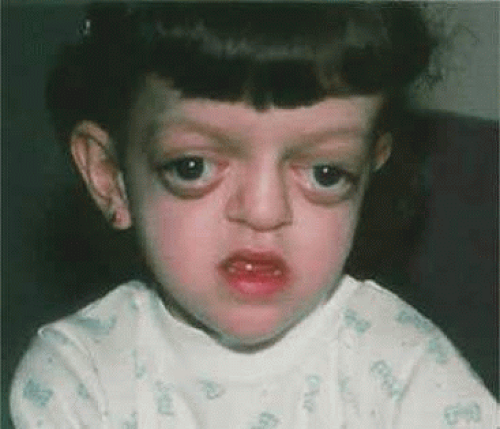 Figure 41.11. Crouzon syndrome showing typical midfacial findings including prominent exophthalmos, hypertelorism, and recessed supraorbital and lateral orbital rims. |
Calvarial suture synostosis in Crouzon syndrome varies, which is why no head shape is characteristic of this syndrome.23 The premature and progressive craniosynostosis is often observed at birth and is usually completed by 2 to 3 years of age.24 Despite this variable presentation, between 96% and 98% of patients with this syndrome display radiographic evidence of fusion of the coronal and sagittal sutures, whereas 79% have lambdoidal synostosis.18 This pattern of fusion leads to the somewhat more predicable skull shape, the features of which include a shortened calvaria, steep forehead, and flattened occiput.3,25,26,27,28,29,30
Early fusion of the cranial sutures imposes physical restraints on the growing brain, which results in neurologic complications. These included headaches (29% to 50%), mental retardation (13%), and seizures (11.5%).7,16,23 In addition to synostosis of the calvarial sutures, the cranial base sutures and midface sutures of the orbits and maxilla display premature fusion.16,31,32,33,34,35,36,37 This may lead to hypoplasia of the ethmoid, frontal, maxillary, and sphenoid bones, which contributes to the midfacial retrusion and shallow, widespread orbits characteristic of the “toadlike” facies associated with Crouzon syndrome.16,30 Other consistent facial features include a parrot-beak nose, hypoplastic cheekbones, flattened nasal bridge, hypoplastic supraorbital ridge, hypertelorism, and shallow orbits with resulting proptosis.24
Hypertelorism and exophthalmos are universal features in Crouzon syndrome.16 Complications arising from the proptosis include exposure keratopathy, exposure conjunctivitis, and more rarely, globe subluxation anterior to the eyelids (Fig. 41.12).15,16 These patients also exhibit exorbitism, which is an increased angle of divergence of the orbits in excess of the normal 90 degrees. Other ocular anomalies that have been sporadically reported in Crouzon syndrome include aniridia, anisocria, blue sclera, cataracts, compound astigmatism, corectopia, ectopia lentis, glaucoma, iris coloboma, keratoconus, megalocornea, microcornea, nystagmus, and optic nerve hypoplasia.30,38,39 Adenexal abnormalities may include canthal dystopia, ectropion, entropion, nasolacrimal duct obstruction, and ptosis.40,41,42,43,44,45,46
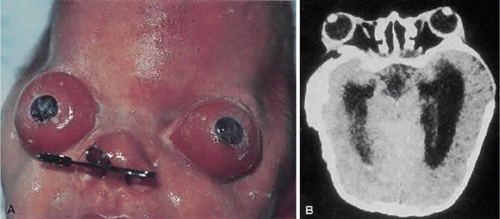 Figure 41.12. A. Child with Crouzon syndrome and severe orbital hypoplasia with resultant proptosis anterior to eyelids. B. Axial computed tomogram of child in Figure 41.12A demonstrating extreme orbital shallowness. |
Strabismus has been described in as many as 92% of patients with Crouzon syndrome, with V-pattern exotropia the most common variety. Esotropia, exotropia, hypertropia, and vertical and horizontal phorias have also been reported.7,23,47,48 The frequently encountered V-pattern exotropia has been postulated to occur secondary to an underaction or absence of the superior oblique muscle with a pseudo-overreaction of the inferior oblique muscle.48 In fact, absent or abnormally inserting extraocular muscles may contribute to the aforementioned patterns for strabismus.49
Optic nerve dysfunction may be present in up to 80% of patients with Crouzon syndrome.23 Optic nerve involvement may present clinically as either optic atrophy or papilledema and its etiology remains elusive. This papilledema, in a study by Bentelson,23 was not correlated to hydrocephalus, increased intracranial pressure, or narrow optic canals.
Nonophthalmic findings in Crouzon syndrome include crowding of the maxillary teeth, lateral palatal swellings, and jaw malocclusion secondary to maxillary hypoplasia.16 Of these patients, 55% acquire conductive hearing loss, whereas 30% have atretic external auditory canals.16 Obstructive sleep apnea, which is common, is related to a constricted nasopharynx and midfacial retrusion.15,16,18 Cervical spine anomalies occur in 30% of patients; most of these abnormalities involve fusion of the C2 and C3 vertebrae.16 Radiographic evidence of calcification of the stylohyoid ligament has also been reported in 88% of patients. As mentioned previously, headaches, seizures, and hydrocephalus may occur; however, mental retardation is rare.16
Apert Syndrome
Apert syndrome (acrocephalosyndactyly type I) was described by Apert in 1906,50 although others had reported similar cases before his publication.50,52 Apert syndrome is characterized by craniosynostosis with symmetric syndactyly of the hands and feet. Two clinical categories of acrocephalosyndactyly exist: typical (or Apert) variant and the atypical variant. Typical acrocephalosyndactyly is clinically distinguishable by complete distal fusion of the soft tissues of digits 2, 3, and 4, with variable degrees of bony fusion in both the hands and feet.53 A middigital hand mass with a common single nail to digits 2 through 4 is present. Despite a range of craniofacial variability, it is the clinical reproducibility of the hand anomaly that distinguishes these individuals from those with the atypical acrocephalosyndactyly.
Apert syndrome has an estimated frequency of 1 per 160,000 live births53 and comprises approximately 4.5% of all cases of craniosynostosis. Although this syndrome is inherited in an autosomal-dominant pattern with complete penetrance, most reported cases are sporadic.54,55,56,57,58 Advanced paternal age appears to be a risk factor for the fresh sporadic mutations53,59,60 and the syndrome has been observed with equal incidence across all populations.7 Apert syndrome appears to be caused by mutations located in a region of the FGFR2 gene.61 In most patients with Apert syndrome, one of two specific mutations in exon IIIa have been found.61,62 Mutations in exon IIIa have also been found to cause Crouzon, Pfeiffer, and Jackson-Weiss syndromes, although these are in a different part of exon IIIa than the mutations that cause Apert syndrome.63
The characteristic limb malformations differentiate Apert syndrome from other craniosynostosis associated with syndactyly.50 Based on craniofacial dysmorphology alone, Apert syndrome can be confused with Crouzon, Pfeiffer and, less often, Saethre-Chotzen syndromes. Despite multiple and variable suture involvement in Apert syndrome, craniosynostosis usually affects the coronal sutures, thereby limiting the anterior and posterior growth of the cranium.15 The skull contour is typically tall, with a steepened forehead and a flattened occiput. Similar to Crouzon syndrome, there is hypoplasia of the midface, hypertelorism, and shallow orbits with proptosis, which may lead to exposure keratopathy. Patients with Apert syndrome also may possess parrot-beak nose, low-set ears, and a horizontal groove above the supraorbital ridge (which diminishes with age) as well as down-slanting (antimongoloid) palpebral fissures (Fig. 41.13).
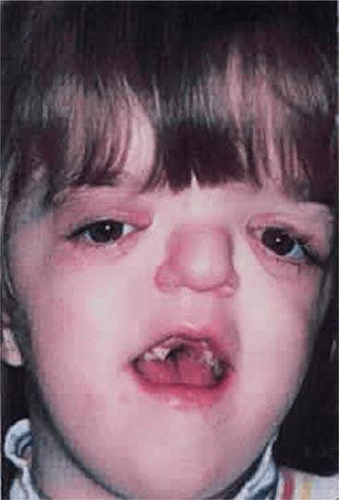 Figure 41.13. Apert syndrome showing frontal overhang, “parrot-beak” nose, mild exophthalmos, and exorbitism. |
The hypertelorism, decreased orbital volume, and proptosis in Apert syndrome are secondary to the midface retrusion and dysmorphic cranial base and are generally less severe than in Crouzon syndrome.15 Strabismus occurs in 70% to 100% of patients and is usually of the exotropic variety. V-pattern exotropia predominates, although V-pattern esotropia may be observed. Absence of the superior rectus muscle, pseudoinferior oblique overaction secondary to excyclotorsion of the orbits, absence of the superior oblique muscle, and structural abnormalities of the extraocular muscles have been reported as well.48,64 Refractive errors, such as high astigmatism and anisometropia, are common in this syndrome. Other rare ophthalmic manifestations include keratoconus, ectopia lentis, congenital glaucoma, and optic atrophy.9,36,37
Apert syndrome does not necessarily include mental retardation, although it is often presented by clinicians to patients’ families in this way.65 Intelligence is often subnormal and, in one study of 15 patients with Apert syndrome, the average IQ was 72.5.66 Development may be adversely affected by progression of increased intracranial pressure, although early craniotomy does not appear to ameliorate development.67 Other neurologic anomalies associated with Apert syndrome include megalocephaly, as well as defects of the corpus callosum, brain, and gyral anomalies.66,68
Oral manifestations of Apert syndrome include crowded teeth, impaction, and thickened gingiva.15,69 Clefting of the soft palate and bifid uvula occur in 30% of patients and malocclusion, usually class 3, related to the maxillary hypoplasia is seen. Many cardiac and visceral anomalies, such as coarctation of the aorta, ventricular septal and atrial septal defects, patent ductus arteriosus, and tracheoesophageal and pyloric stenosis have been reported, but such findings are relatively uncommon.70 In adolescent patients, 70% display peculiar acneiform eruptions on the trunk and forearms.71 Conductive hearing loss, often from chronic otitis media, is extremely common with the incidence of deafness reported to be as high as 30%.72,73,74
Syndactyly of the hands and feet is a major feature of Apert syndrome (Fig. 41.14). As mentioned previously, a middigital hand mass involving the second, third, and fourth digits is observed. The first and fifth digits may be adjoined, but when the thumb is separate it is usually broad. Symmetric syndactyly of the toes is seen, with medial deviation of the great toe and progressive fusion of the midfoot and hindfoot in the supinated position.75 Radiologic studies show some degree of brachydactyly in all five digits, synphalangism of the interphalangeal joints, and eventual fusion of the carpal, tarsal, and interphalangeal joints over time.7,76 Other orthopedic anomalies in Apert syndrome include ankylosis of the elbow, shoulder, and hip and cervical stenosis.77
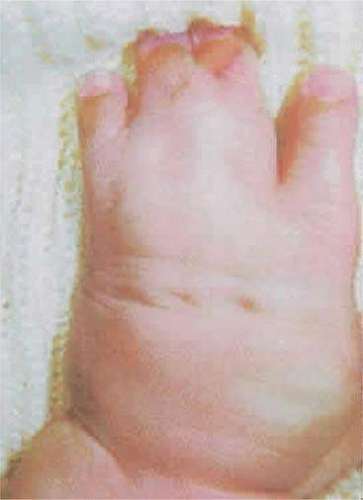 Figure 41.14. Hand of child with Apert syndrome showing proximal syndactyly of digits 2 and 3, 4, and 5 and near-total fusion of digits 3 and 4. (From Fries PD, Katowitz JA: Congenital craniofacial anomalies of ophthalmic importance. Surv Ophthalmol 35:87, 1990, with permission from Elsevier Science.) |
Several additional cranial synostoses share common craniofacial features with Crouzon syndrome but occur less frequently; these include Carpenter, Pfeiffer, and Sathre-Chotzen syndrome.
Carpenter Syndrome
Carpenter syndrome (acrocephalopolysyndactyly type II) is a rare entity first described by Carpenter in 1901. It was finally recognized as a distinct entity in 1966 when Tentamy reported 1 case and studied 12 cases previously described in the literature under the name of Laurence-Moon-Biedl syndrome.78,79 Carpenter syndrome is clinically characterized by craniosynostosis, preaxial polydactyly of the feet, variable brachydactyly, syndactyly, and obesity. Inheritance is autosomal recessive, but many cases represent new spontaneous mutations and it has been associated with mutations in the RAB23 gene located on chromosome 26.72,79
Ocular findings are less distinct owing to the rarity of the disorder. Hypertelorism with relative telecanthus and epicanthal folds has been described in most cases.80 Shallow orbits with proptosis and mild down-slanting of the palpable fissure may also be observed. Corneal opacity, microcornea, optic atrophy, and pseudopapilledema occur sporadically.78,79,80,81
The craniosynostosis of Carpenter syndrome is characterized by multisutural involvement. Variable premature fusion of the coronal, lambdoidal, and sagittal sutures normally result in a grossly normal calvaria, which is usually shortened in all directions.15,79,82 Most affected patients are mentally retarded; however, those with normal intelligence have been reported.82,83 Short stature and obesity are usually present. Between 30% and 50% of patients have congenital heart defects, including atrial and ventricular septal defects, patent ductus arteriosus, and tetralogy of Fallot.79,84
Patients with Carpenter syndrome may also have low-set ears, a hypoplastic mandible, flattened nasal bridge, and high arched palate.7,80,81 Other anomalies include pes varus, genu valgum, laterally displaced patella, ilial flaring, and decreased hip mobility.15,74,79,82,83,84,85 Preaxial polysyndactyly of the feet, soft tissue syndactyly of the hands, and brachydactyly and duplication of the second toe may be seen in many of these patients.
Pfieffer Syndrome
The combination of craniosynostosis, broad thumbs and toes, and partial soft tissue syndactyly of the fingers and toes was described by Rudolf Pfieffer86 in 1964 in eight members of one family affected over three generations (Fig. 41.15). Pfeiffer syndrome is an autosomal-dominant disorder with complete penetrance and variable expression.87,88 The genetic mutations in Pfeiffer syndrome have been identified as FGF2 on chromosome 10 and FGF1 on chromosome 8.89 Interestingly, identical mutations of FGF2 have been identified in Crouzon and Pfieffer syndrome despite their phenotypic differences in appearance. Cohen87 classified Pfeiffer syndrome into three subtypes:
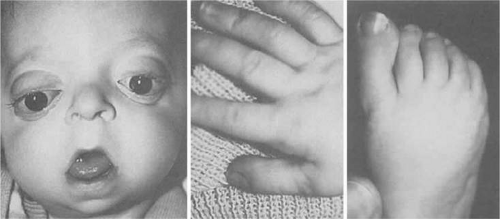 Figure 41.15. A. Clinical photograph of Pfeiffer syndrome demonstrating exophthalmos, hypertelorism, small mandible, “parrot-beak” nose, recessed supraorbital ridges, and increased forehead height. B. Enlarged thumb of same patient. C. Broad, long great toe. (From Fries PD, Katowitz JA: Congenital craniofacial anomalies of ophthalmic importance. Surv Ophthalmol 35:87, 1990, with permission from Elsevier Science.) |
Patients with type I have normal intelligence, whereas types II and III have developmental retardation. The usefulness of this classification lies chiefly in genetic counseling, given that much clinical overlap exists among the different subtypes.
The ophthalmic manifestations in Pfeiffer syndrome closely parallel those found in Crouzon and Apert syndromes. Hypertelorism, ocular proptosis, oxybrachycephaly (often secondary to bilateral coronal synostosis), strabismus, flattened nasal bridge with beaked nose, maxillary hypoplasia with a relative mandibular prognathism, and malocclusion are typical findings in this syndrome. Other abnormalities include high arched palate, crowded teeth, hydrocephalus, seizures, bifid uvula, low-set ears, and conductive hearing loss.90
The distinguishing features of this syndrome are the broad, medially deviated great toes and thumbs. In fact, the ratio of the width of the great toe to the second toe may be useful in diagnosing this syndrome.87,91 Brachydactyly, clinodactyly, and partial soft tissue syndactyly may be present, whereas radiographic studies of the hand and foot show the occasional absence of the middle phalanges, broad distal thumb phalanges, triangulation of the proximal thumb, and the unusual appearance or the duplication of the first metatarsal bone.92 Associated abnormalities include lumbar hyperlordosis, fusion of lumbar or cervical vertebrae, shortened humerus, umbilical hernia, preauricular skin tags, and supernumerary teeth.
Saethre-Chotzen Syndrome
Saethre-Chotzen syndrome is characterized by craniosynostosis, low-set frontal hairline, facial asymmetry, deviated nasal septum, and partial simple syndactyly, especially involving the middle and index fingers (Fig. 41.16).93,94 This syndrome, as described by Saethre and Chotzen, is an autosomal-dominant disorder with high penetrance and variable expression. The mutation that causes Saethre-Chotzen syndrome has been identified. As with the FGFR craniosynostotic disorders, identification of the mutated gene was accomplished by analysis of candidate genes in the region of a chromosome identified by linkage analysis. In the case of Saethre-Chotzen syndrome, linkage analysis mapped the disease locus to 7p21–22.95,96 A basic helix-loop–helix transcription factor called TWIST had been previously localized to the same region of chromosome 7, and several different types of mutations in the TWIST gene were found in patients with Saethre-Chotzen syndrome.96,97 Paternal age is a risk factor for new mutations and translocation defect of chromosome 7 have been noted in certain families with the syndrome.7,97,98,99
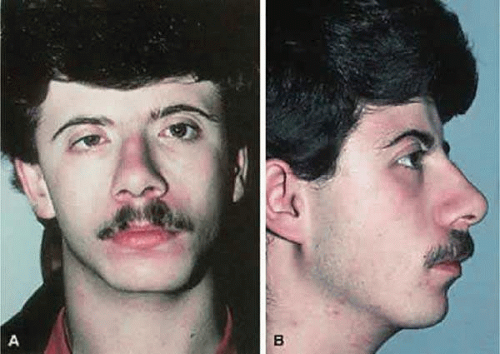 Figure 41.16. Frontal (A) and lateral (B) view of patient with Saethre-Chotzen syndrome with low-set hairline, ptosis, strabismus, mild facial asymmetry, and low-set ears. (A: from Fries PD, Katowitz JA: Congenital craniofacial anomalies of ophthalmic importance. Surv Ophthalmol 35:87, 1990, with permission from Elsevier Science.) |
Saethre-Chotzen syndrome is often underdiagnosed because the craniosynostosis is diagnostic for the syndrome and cosmetic abnormalities are usually mild. Variability in head shape is usually seen as plagiocephaly. Scaphocephaly, brachycephaly, and oxycephaly have also been reported.100,101 The midface is spared the retrusion seen in the other craniosynostosis. Frontal bossing with a high forehead, which is often slightly concave, and low-set frontal hairline are fairly distinctive features. The ears are small, misshapen, and often set low.102 Hearing impairment has also been observed. Dental anomalies occur in approximately 50% of patients; they include malocclusion, high-arched palate, and cleft palate.102,103,104,105
Although most patients with Saethre-Chotzen syndrome have normal intelligence, seizures and schizophrenia have been reported.7,93,94 Hand anomalies are frequent and most commonly include simple partial syndactyly, clinodactyly, and simian creases.101
Ophthalmic features of Saethre-Chotzen differ from the other craniosynostoses. Ptosis is far more common in Saethre-Chotzen and strabismus is seen in over 50% of cases.100,101 Proptosis and hypertelorism are less common than in Apert and Crouzon syndromes, presumably because of the relative absence of midface hypoplasia. Nasolacrimal drainage abnormalities with epiphora occur in 50% of patients, whereas optic atrophy and amblyopia are present in approximately 24%.101
Clefting Syndromes
The second major category of craniofacial abnormalities involves the clefting syndromes. As a group, craniofacial clefting is both etiologically and pathogenetically heterogeneous.106,107 The meaning of the word cleft is easily understood when faced with a clinical example such as a cleft lip or palate. However, defects in opposition of junctional structures are not always so obvious and easy to classify.
Historically, many attempts have been made to classify the clefting and craniofacial anomalies. Efforts put forth by Morin et al.,108 Lund,109 Burian,110 Sanvenero-Rosselli,111 Franceschetti et al.,112 and Tessier113 have enjoyed only brief popularity, because there were numerous shortcomings and overlaps, as well as a poor understanding of the underlying pathogenesis.
Paul Tessier, in 1976, presented an orderly classification of the clefting syndromes that has been universally accepted because of its simplification of nomenclature106,113 (Fig. 41.17). Tessier’s system assigns each craniofacial cleft a number from 0 to 14, beginning at 0 in the midline of the lower face and progressing clockwise around the right orbit ending at 14 in the upper facial midline. The left side of the face is organized in the same manner, except the designation numbers run counterclockwise. Tessier’s classification scheme is purely descriptive and provides no pathogenetic or etiologic information. Nevertheless, this is the most widely accepted system because it facilitates recording and communication among observers.106,113
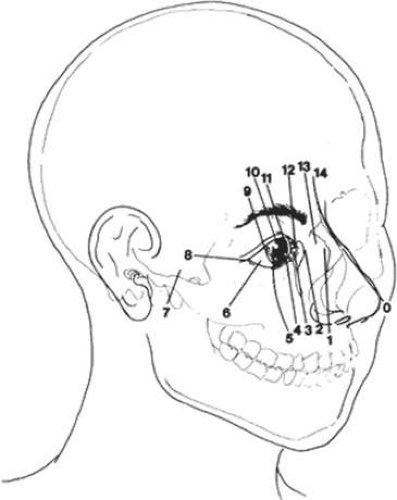 Figure 41.17. Oblique view of skull with adnexal soft tissue demonstrating positions of facial clefts. (From Fries PD, Katowitz JA: Congenital craniofacial anomalies of ophthalmic importance. Surv Ophthalmol 35:87, 1990, with permission from Elsevier Science.) |
The morphogenesis of facial clefting is not well understood and is a topic of much debate. To explain the clefting phenomenon, Dursy in 1869114 and His in 1892115 proposed the classic theory of fusion. They suggested that clefts develop as a result of the failure of fusion of the normal embryonic facial processes. In contrast to the classic theory, Johnston116 and Wetson117 subsequently implicated either (a) the failure of neural crest cell migration, which prevents the mesenchyme responsible for skeletal and connective tissue formation of the face or (b) the degeneration of these cells before their migration as the source of clefts. More recently, studies by Vermiej-Keers et al.118,119 suggests that neural crest cell migration may not be the source of all facial clefting. The group led by Vermiej-Keers identified four primary sites in embryonic facial development that may lead to facial clefts: (a) between the lateral and medial facial processes, (b) between the lateral nasal and maxillary processes, (c) between the maxillary and mandibular processes, and (d) between the palatine processes.119 These sites are identified as true clefts because they are associated with normal sutures of embryologic development. True clefts tend to be well-defined and occur within the seventh week of gestation.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


